{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
薄荷内生细菌的多样性及组成分析
[陈泽斌1, 2  , 李冰
, 李冰3, * , 王定康4 , 余磊1 , 徐胜光1 , 张永福1 , 任禛1 ]
, 李冰, 王定康|
|


作者简介: 陈泽斌(1985—),男,云南昆明人,博士,副教授,主要从事环境微生物的教学及科研工作。E-mail:zbchenkmu@163.com
为了调查薄荷内生细菌种类多样性,应用高通量测序技术测定薄荷内生细菌的16S rDNA-V4变异区序列,采用Qiime和Mothur等软件整理和统计样品序列数目和操作分类单元(OTUs)数量,分析物种的丰度、分布和α多样性,以及物种丰富度的差异。研究获得用于分析的有效序列和OTU数为60 034/226;稀疏曲线表明测序深度充分,OTU的数量接近于饱和。薄荷(样品名BH)的Chao 1指数为226.0,Shannon多样性指数为2.538。薄荷内生细菌分布于以下10个属:食酸菌属( Acidovorax,65.34%)、鞘氨醇单胞菌属( Sphingomonas,17.32%)、土地杆菌属( Pedobacter,5.88%)、甲基杆菌属( Methylobacterium,4.33%)、 Luteolibacter属(1.94%)、土壤杆菌属( Agrobacterium,1.53%)、鞘氨醇杆菌属( Sphingobacterium,1.30%)、金黄杆菌属( Chryseobacterium,0.92%)、黄杆菌属( Flavobacterium,0.78%)、 Dyadobacter属(0.67%);薄荷内生细菌优势菌属为食酸菌属( Acidovorax,65.34%)和鞘氨醇单胞菌属( Sphingomonas,17.32%)。Illumina MiSeq高通量测序技术为植物内生细菌的研究提供了更加准确、科学的数据资源。
In order to investigate the endophytic bacterial community, leaves samples were collected from Menthae Haplocalycis Herba. The 16S rDNA-V4 region of leaves collected from Menthae Haplocalycis Herba were sequenced by Illumina MiSeq high-throughput sequencing technology. Using Qiime and Mothur softwares, the number of sequences and operational taxonomic units (OTUs) for sample was sorted and calculated, the species abundance and distribution, Alpha diversity index and difference in species abundance among samples were analyzed. The number of effective sequences and OTUs for each sample were 60 034/226. The rarefaction curves showed that adequate sampling was achieved. The number of OTUs was close to saturation. The chao 1 and Shannon indexes of sample BH were 226.0 and 2.538, respectively. The species of endophytic bacteria in Menthae Haplocalycis Herba belonged to Acidovorax (65.34%), Sphingomonas (17.32%), Pedobacter (5.88%), Methylobacterium (4.33%), Luteolibacter (1.94%), Agrobacterium (1.53%), Sphingobacterium (1.30%), Chryseobacterium (0.92%), Flavobacterium (0.78%) and Dyadobacter (0.67%). The dominant species were Acidovorax and Sphingomonas with a percentage of 65.34% and 17.32% respectively. Illumina high-throughput sequencing technology provided more accurate and scientific data resources for the study of endophytic bacteria in Menthae Haplocalycis Herba.
现已从多种健康植物的根、茎、叶、果实、种子中分离出革兰氏阳性菌和革兰氏阴性菌的植物内生细菌, 共达129种(隶属于54个属)[1]。目前研究比较系统的植物有棉花、小麦和花生。优势种类主要分布于假单胞属(Pseudommonas)、芽孢杆菌属(Bacillus)、肠杆菌属(Enterobacter)、土壤杆菌属(Agrobacterium)4个属[2]。目前, 还没有发现不存在内生细菌的植物种类或器官。
薄荷, 土名“ 银丹草” , 为唇形科植物, 即同属其他干燥全草, 多生于山野湿地河旁, 根茎横生地下, 多生于2 100 m海拔高度, 但也可在3 500 m海拔上生长, 是一种有特种经济价值的芳香作物[3]。全株清气芳香, 叶对生, 花小淡紫色, 唇形, 花后结暗紫棕色的小粒果。薄荷是中华常用中药之一。它是辛凉性发汗解热药, 治流行性感冒、头疼、目赤、身热、咽喉、牙床肿痛等症[4]。外用可治神经痛、皮肤瘙痒、皮疹和湿疹等。光棚温室采摘的薄荷是春节餐桌上的鲜菜, 清爽可口; 平常以薄荷代茶, 清心明目。在中国, 薄荷主要以江苏、安徽两省产量最大[5]。
目前, 关于薄荷内生菌多样性的研究均采用传统培养方法[6, 7], 采用非培养方法对其进行研究还未见报道。而许多研究已经证实, 通过传统的分离培养方法鉴定的微生物只占环境生物总数的0.1%~10%[8], 因此有必要采用非培养方法对薄荷内生细菌的种类组成进行研究。本研究采用近年来兴起的Illumina MiSeq第二代测序技术全面而准确地对薄荷内生细菌种类组成进行研究, 相较于传统的纯培养方法及以16S rDNA为基础的非培养方法, 能够产生测序覆盖深度更大的数据量, 检测到传统纯培养和非培养技术未能发现的低丰度植物内生细菌种类, 为丰富植物微生态学理论及基因工程菌的研究和应用奠定基础。
薄荷(样品编号BH)样品取自昆明学院农学院果蔬实践园, 取样时选择生长性状良好、无病害症状的健康叶片, 取样后立即放入样品袋中, 低温保鲜, 12 h内处理[9]。
植物基因组DNA快速提取试剂盒DP305为天根生化科技(北京)有限公司产品; GeneJET胶回收试剂盒为美国Thermo Scientific公司产品; 引物合成由生工生物工程(上海)股份有限公司完成; 高保真PCR酶Phusion® High-Fidelity PCR Master Mix with GC Buffer、Illumina建库试剂盒NEB Next® UltraTM DNA Library Prep Kit为New England Biolabs公司产品; MiSeq测序试剂盒v2、高通量测序仪(MiSeq System SY-410-1003)为美国Illumina公司产品; 分光光度计(Nano drop 2000/2000C)为美国Thermo Scientific公司产品。
将薄荷叶片用自来水冲洗干净, 再用75%乙醇浸泡1 min, 无菌水冲洗3次, 用无菌滤纸吸干表面水分, 参考文献[10]检验表面消毒效果。
按参考文献[11]的方法提取薄荷叶片总DNA, 并用0.8%琼脂糖凝胶电泳检查DNA的纯度和浓度, 取适量样品于离心管中, 使用无菌水稀释样品至1 ng· μ L-1。
以稀释后的基因组DNA为模板, 使用带Barcode的16S rDNA-V4区特异引物515F(5’ -GTTTCGGTGCCAGCMGCCGCGGTAA-3’ )和806R(5’ -CAGATCGGACTACHVGGGTWTCTAAT-3’ ), 使用高效和高保真酶(New England Biolabs公司的Phusion® High-Fidelity PCR Master Mix with GC Buffer)进行PCR, 确保扩增效率和准确性。
PCR产物使用2%琼脂糖凝胶进行电泳检测; 根据PCR产物浓度进行等浓度混样, 充分混匀后使用2%琼脂糖凝胶电泳检测PCR产物, 使用Thermo Scientific公司的GeneJET胶回收试剂盒回收产物。
1.7.1 构建文库
使用New England Biolabs公司的NEB Next® UltraTM DNA Library Prep Kit for Illumina建库试剂盒进行文库的构建。通过3'-5'核酸外切酶及聚合酶的共同作用, 修复带有突出末端的DNA片段。在修复平整的DNA片段3'端引入单碱基“ A” , 接头的3'末端含有单碱基“ T” , 从而保证DNA片段和接头能够通过“ A” “ T” 互补配对连接, 并防止接头连接DNA片段的过程中, DNA片段彼此相连。在连接酶的作用下, 孵育含有标签的接头与DNA片段, 使其相连。利用PCR选择性地富集两端连有接头的DNA片段, 同时扩增DNA文库。PCR应尽量使用较少的循环数, 避免PCR扩增中文库出现错误。构建好的文库经过Qubit定量和文库检测, 合格后, 使用MiSeq进行上机测序[12, 13, 14, 15]。
1.7.2 均一化文库
将样品DNA文库均一化至10 nmol· L-1后等体积混合。
1.7.3 上机测序
将混合好的文库(10 nmol· L-1)逐步稀释定量至4~5 pmol· L-1后用Illumina MiSeq测序仪测序, 进行生物信息分析。本研究的测序和生物信息服务在北京诺禾致源生物信息科技有限公司完成。
测序得到的原始数据(raw data), 存在一定比例的干扰数据(dirty data), 为了使信息分析的结果更加准确、可靠, 首先对原始数据经过拼接、过滤、去嵌合体后, 得到有效数据(effective tags)[16], 剔除宿主叶绿体和线粒体序列, 然后对有效数据在97%水平上进行OUT聚类, 并利用Greengene数据库进行物种注释[17]。通过对OTUs进行丰度、α 多样性、β 多样性以及物种在各个分类水平上的群落结果统计分析, 得到微生物群落结构组成[18, 19]。
利用uparse(version 7.1 http:∥qiime.org/)软件平台进行OTU聚类, 采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析, 并在各个水平上统计每个样品的群落组成; 利用Mothur软件做稀释曲线分析; 分别利用香农指数(Shannon)、辛普森指数(Simpson)和物种丰富度指数(ACE)公式计算细菌生态多样性指数; 利用Excel和 Mothur软件制作Venn图; 利用Fasttree软件+R语言制作热图; 利用诺禾致源自主开发的软件SVG制作特定物种分类树图。
BH样品所测得的60 731条PE Reads长度分布在190~350 bp范围内, 长度为250 bp的序列最多, 有60 217条。从序列长度的分布来看, 与16S rDNA-V4区序列长度大致吻合。
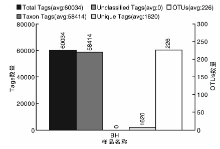
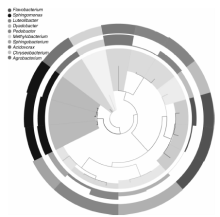
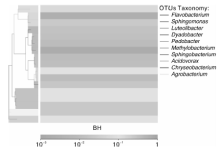
BH样品共获得60 034条Tags(过滤后得到的拼接序列数), 可分为226个OTUs(97%的序列相似性, 下同), 包括58 414条可获得分类信息的Tags, 1 620条低频Tags(图1)。BH可以注释到14 424个属。图2展示了BH样品中相对丰度排名前10个属细菌所对应的OTUs的系统发生关系数据, 表明薄荷内生细菌主要分布于以下10个属:黄杆菌属(Flavobacterium)、鞘氨醇单胞菌属(Sphingomonas)、Luteolibacter属、Dyadobacter属、土地杆菌属(Pedobacter)、甲基杆菌属(Methylobacterium)、鞘氨醇杆菌属(Sphingobacterium)、 食酸菌属(Acidovorax)、金黄杆菌属(Chryseobacterium)、 土壤杆菌属(Agrobacterium); 从OTUs丰度聚类图来看(图3), 在属的分类水平上, 相对丰度由大到小依次为:食酸菌属(Acidovorax)> 鞘氨醇单胞菌属(Sphingomonas)> 土地杆菌属(Pedobacter)> 甲基杆菌属(Methylobacterium)> Luteolibacter属> 土壤杆菌属(Agrobacterium)> 鞘氨醇杆菌属(Sphingobacterium)> 金黄杆菌属(Chryseobacterium)> 黄杆菌属(Flavobacterium)> Dyadobacter属。
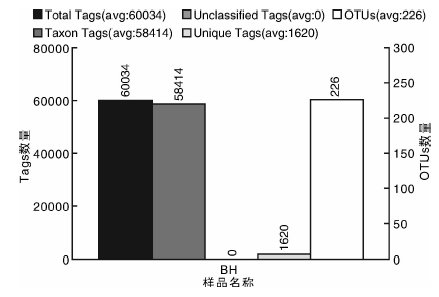 | 图1 样品的Tags和OTUs数目统计Fig.1 The statistics of Tags and OTUs number in sample BH |
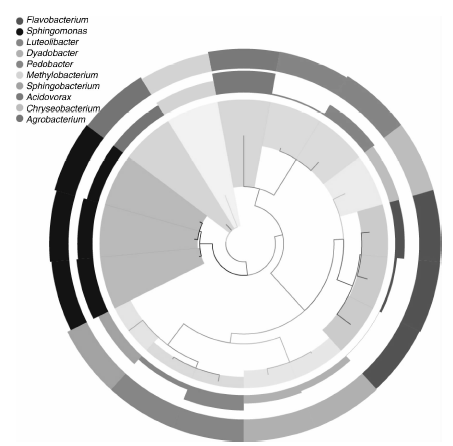 | 图2 OTUs的系统发育关系及其物种注释Fig.2 Phylogenetic relationship species annotation of OTUs |
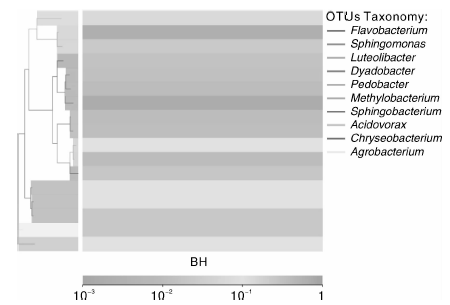 | 图3 OTUs丰度聚类图Fig.3 Abundance clustering figure of OTUs |
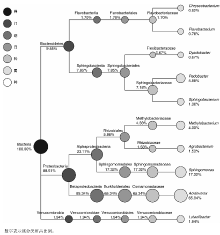
从门的分类水平来看(图4), 薄荷内生细菌在拟杆菌门(Bacteroidetes, 9.55%)、变形菌门(Proteobacteria, 88.51%)、疣微菌门(Verrucomicrobia, 1.94%)中均有分布; 从纲的分类水平来看(图4), 薄荷内生细菌在Flavobacteriia纲(1.70%)、Sphingobacteriia纲(7.85%)、α -变形菌纲(Alphaproteobacteria, 23.17%)、β -变形菌纲(Betaproteobacteria, 65.34%)、疣微菌纲(Verrucomicrobiae, 1.94%)中均有分布; 从目的分类水平来看(图4), 薄荷内生细菌在黄杆菌目(Flavobacteriales, 1.70%)、鞘脂杆菌目(Sphingobacteriales, 7.85%)、根瘤菌目(Rhizobiales, 5.86%)、鞘脂单胞菌目(Sphingomonadales, 17.32%)、伯克氏菌目(Burkholderiales, 65.34%)、疣微菌目(Verrucomicrobiales, 1.94%)中均有分布; 从科的分类水平来看(图4), 薄荷内生细菌在黄杆菌科(Flavobacteriaceae, 1.70%)、屈挠杆菌科(Flexibacteraceae, 0.67%)、鞘脂杆菌科(Sphingobacteriaceae, 7.18%)、甲基杆菌科(Methylobacteriaceae, 4.33%)、根瘤菌科(Rhizobiaceae, 1.53%)、鞘脂单胞菌科(Sphingomonadaceae, 17.32%)、丛毛单胞菌科(Comamonadaceae, 65.34%)、疣微菌科(Verrucomicrobiaceae, 1.94%)中均有分布; 从属的分类水平来看(图4), 薄荷内生细菌在食酸菌属(Acidovorax, 65.34%)、鞘氨醇单胞菌属(Sphingomonas, 17.32%)、土地杆菌属(Pedobacter, 5.88%)、甲基杆菌属(Methylobacterium, 4.33%)、Luteolibacter属(1.94%)、土壤杆菌属(Agrobacterium, 1.53%)、鞘氨醇杆菌属(Sphingobacterium, 1.30%)、金黄杆菌属(Chryseobacterium, 0.92%)、黄杆菌属(Flavobacterium, 0.78%)、Dyadobacter属(0.67%)中均有分布; 由此可见, 食酸菌属(Acidovorax, 65.34%)和鞘氨醇单胞菌属(Sphingomonas, 17.32%)为薄荷内生细菌的优势种群。
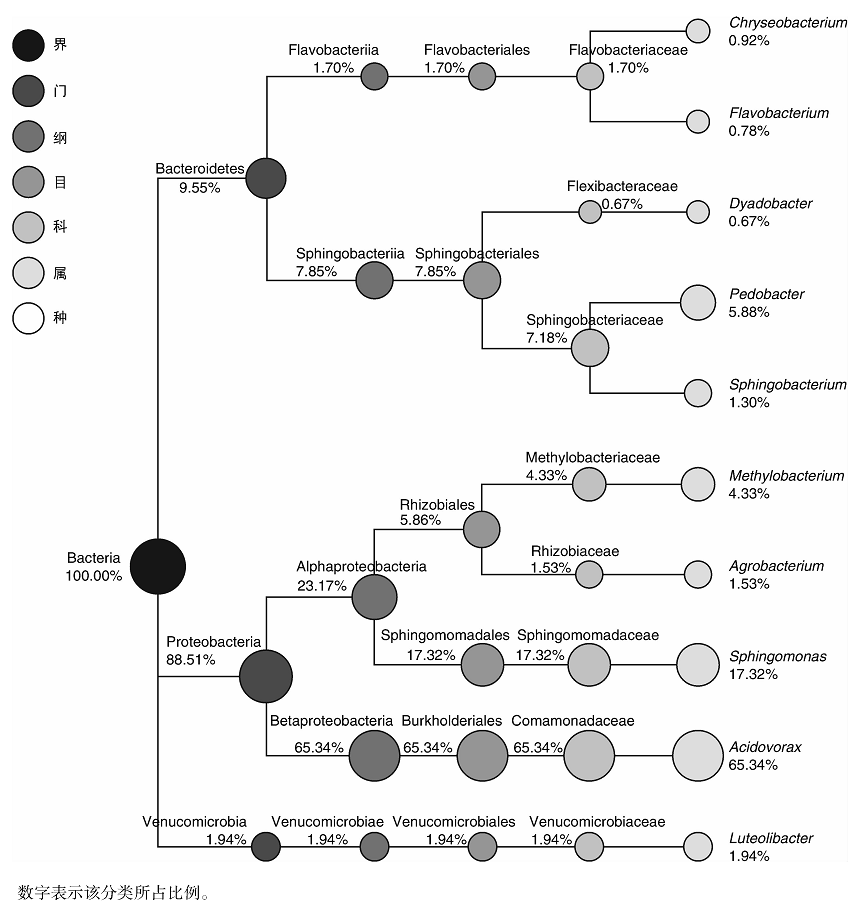 | 图4 样品BH中特定物种分类树Fig.4 Classification tree of particular species from sample BH |
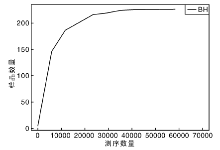
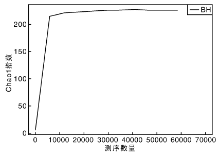
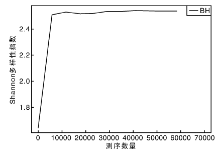
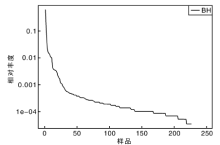
从稀释曲线来看(图5), 随着测序数量的增加, 稀释曲线斜率逐渐降低, 趋向平坦, 进入平台期, 说明再增加测序数量也只会产生少量新的OTUs; 从Chao1指数曲线来看(图6), Chao1指数在8 000的测序数量范围内增幅最大, 在8 000以后曲线斜率下降; 从Shannon指数曲线来看(图7), Shannon指数在8 000的测序数量范围内增幅最大, 在8 000以后曲线斜率下降。3条曲线在测序数量超过8 000后都趋近平缓, 说明测序数据量足够大, 可以反映样品中绝大多数的微生物信息; 从Rank Abundance曲线来看(图8), BH样品在水平方向的跨度较大, 在垂直方向的平滑程度高, 说明薄荷内生细菌的丰度大, 且物种的分布较均匀。α 多样性分析表明, 薄荷(样品名BH)的Chao 1指数为226.0, Shannon多样性指数为2.538。
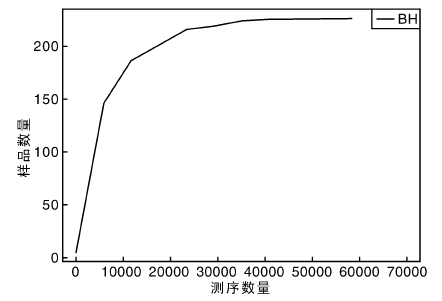 | 图5 稀释曲线Fig.5 Rarefaction curve |
 | 图6 Chao1指数曲线Fig.6 Chao1 indexes curve |
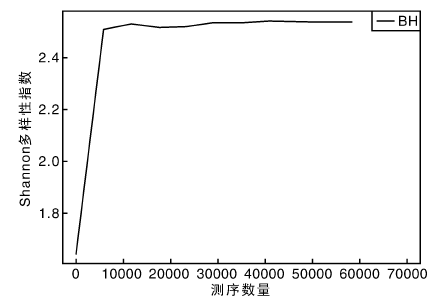 | 图7 Shannon多样性指数曲线Fig.7 Shannon indexes curve |
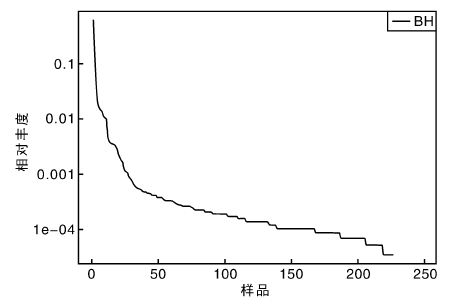 | 图8 Rank abundance曲线Fig.8 Rank abundance curve |
16S rRNA基因很早就应用在微生物菌种鉴定上, 但利用高通量测序进行16S rRNA分析最早开始于2006年, Sogin等[20]应用该技术对深海微生物群落进行了分析, 随后该技术在肠道微生物领域得到了广泛应用[21]。目前该技术应用于植物内生微生物的研究尚未见报道。本研究首次将近几年迅速发展并成为主流的Illumina MiSeq第二代测序技术应用于植物内生细菌研究, 克服了传统分子生物学方法通量低的缺陷, 从基因组水平上解析微生物群落结构, 突破了很多厌氧内生微生物尚不能被分离培养的技术瓶颈[22, 23], 相较于传统的纯培养和以16S rDNA为基础的非培养方法, Illumina MiSeq第二代测序技术可以检测到以往没有检测到、同样扮演着重要角色的低丰度植物内生细菌种类, 丰富植物内生细菌资源, 例如Dyadobacter属(0.67%)。本研究对植物中好氧、厌氧和兼性厌氧内生微生物群落的种类、丰度和分布进行了分析, 显示出高通量技术在植物内生微生物研究中的明显优势, 相较于传统分子生物学的研究结果, 高通量测序技术覆盖了整个内生微生物群落的信息, 能更准确地反映植物内生微生物中不同丰度菌群的组成和真实比例, 本研究可为今后其他研究者使用该技术开展同类研究提供参考。
就植物组织而言, 叶绿体和线粒体与细菌在系统发育上具有高度的同源性[24]。在对薄荷内生细菌的16S rDNA-V4变异区序列进行Illumina MiSeq高通量测序后, 确实发现部分序列归属于宿主叶绿体和线粒体, 存在一定比例的宿主污染现象, 为了使细菌多样性分析结果更加准确、可靠, 本研究剔除了宿主叶绿体和线粒体序列。只选择16S rDNA-V4区作为测序区域, 若要将高通量测序技术更好地应用于植物内生微生物研究, 下一步应在不同高通量测序平台上针对16S rDNA不同可变区的选择及组合做尝试, 评估样品的复杂程度以及宿主的污染情况, 选择合理的测序区域或设计特异性更高的引物, 制定更好的测序策略, 以避免线粒体和叶绿体的干扰。
本研究结果只能将内生细菌注释到属的分类水平, 而第一代Sanger测序可以将细菌注释到种的水平, 这是因为16S rRNA总长约1 540 bp, 包含9个可变区, 第一代测序读长长、准确率较高, 所以能根据测得的全长16S rRNA序列注释到种, 但第一代Sanger测序通量较低, 大规模测序成本高。Illumina MiSeq高通量测序技术, 一般每个样品可以保证测定4~6万个序列数, 由于覆盖深度非常大, 对物种多样性的分析十分有利, 但是通量的增加是以牺牲序列读长为代价, 由于高通量测序读长短, 不可能将16S rRNA的9个可变区全部测序, 所以往往只选择1~3个可变区作为测序区域, 单端测序长度可达到250 bp, 双端测序长度可达到500 bp, 测序片段越短, 在后续对序列进行物种注释时的分辨度越低。
本研究发现, 薄荷内生细菌的优势种群为食酸菌属(Acidovorax, 65.34%)和鞘氨醇单胞菌属(Sphingomonas, 17.32%)。这与前人通过培养方法得出植物优势类群为芽孢杆菌属(Bacillus)细菌的结果不同[25], 这是因为培养方法分析的细菌只限于那些能够在人工培养基上生长的细菌, 而许多研究已经证实, 通过传统的分离培养方法鉴定的微生物只占环境生物总数的0.1%~10%[22, 23], 因此通过传统培养方法得到的结果并不能真实地反映出内生细菌多样性, 这就使得用传统方法研究植物内生细菌的种群结构、多样性及与宿主之间的相互作用受到了极大的限制。Illumina MiSeq高通量测序技术可为植物内生细菌的研究提供了更加准确、科学的数据资源。已有研究表明, 鞘氨醇单胞菌属细菌能够利用的底物广泛, 从多环芳烃类化合物、聚乙烯醇等高聚物到简单无机物N2+都能利用, 某些种属还能产生有价值的生物高分子(如β -胡萝卜素、结冷胶)[26]。说明薄荷内生细菌可为复杂有机物的降解提供良好的微生物来源; 还有研究表明, 定殖生长于植物组织中的内生细菌, 有些能够产生细胞壁降解酶类[27]。这些物质对植物抵抗病原菌侵入、潜伏、扩展蔓延等都是非常重要的。真菌细胞壁的主要组成成分是葡聚糖和几丁质, 某些内生细菌产生细胞壁降解酶如葡聚糖酶 (β -glucanse)和几丁质酶 (Chitinase), 使真菌细胞水解或死亡, 从而达到防病效果。例如, 一种用于防治棉花黄萎病的内生菌, 可产生某些蛋白酶降解毒素达到防病的效果[27]。因此, 推测薄荷内生细菌的优势类群鞘氨醇单胞菌属细菌能够降解某些病原菌的细胞壁或毒素, 与寄主植物的抗病性直接相关。食酸菌属作为内生细菌的优势种群尚属首次报道, 关于食酸菌属细菌的功能、代谢的研究目前还未见报道。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|