

{kind=link}
{kind=link}
{kind=link}
凡纳滨对虾总RNA完整性的正确评估
[易乐飞 , 穆亮亮, 仲改]
, 穆亮亮, 仲改]
, 穆亮亮, 仲改]
|
|


作者简介:易乐飞(1975—),男,湖北荆门人,硕士,副教授,从事水产生物分子遗传学研究。E-mail: yilf@hhit.edu.cn
凝胶电泳技术通常被用于总RNA完整性检测,一般认为28S和18S rRNA条带亮度的比值大于等于2表示总RNA完整性良好,该比值越小表明总RNA降解越严重。为了检测这一标准在水产虾蟹类中是否继续适用,分别对凡纳滨对虾rRNA和mRNA的完整性进行了分析。用TRIzol分离纯化的凡纳滨对虾总RNA经凝胶电泳检测,发现其28S:18S rRNA的比值远小于2;但是以同样的总RNA为模板进行RT-PCR,能顺利扩增出长约1 100 bp的 ACT和 eEF1 A基因序列。进一步的3':5'分析显示这2个内参基因mRNA的3':5' ratio分别为2.79和1.53,直接表明被测mRNA完整性良好。因此,凝胶电泳低估了水产虾蟹类总RNA的完整性,建议采用3':5'分析技术对水产虾蟹类总RNA完整性进行检测。
The denaturing agarose gel electrophoresis and ethidium bromide staining is the most common method used to assess the integrity of total RNA. The 28S rRNA band is approximately twice as intense as the 18S rRNA band after running an intact total RNA sample on a denaturing gel. If the 28S:18S rRNA ratio is less than 2, it is likely that the RNA sample suffered degradation during preparation. The less the ratio is, the more is the degradation. In order to check whether it works for agarose gel analysis to assess the integrity of total RNA extracted from Penaeus vannamei, the integrity of rRNA and mRNA was assessed using two different methods, gel electrophoresis and 3':5' assay in this paper. Total RNA was extracted from Penaeus vannamei with TRIzol reagent and treated by DNase I. A very low value of 28S:18S rRNA ratio, far less than 2, was observed using agarose gel analysis, which suggested the degradation of total RNA sample during preparation. However, using the same total RNA as template, two genes of about 1 100 bp were successfully amplified by reverse transcription PCR. Furthermore, a 3':5' assay was carried out based on the same total RNA. The 3':5' assay was independent of the rRNA integrity and could assess the mRNA integrity directly. A reverse transcription reaction was primed using oligo (dT), and a real time quantitative PCR assay was used to quantitate the levels of 3' and 5' target sequences from ACT and eEF1 A mRNA. The 3':5' ratio of ACT and eEF1 A mRNA were 2.79 and 1.53, respectively, which indicated the high integrity of mRNA. So gel electrophoresis of RNA underestimated the integrity of total RNA. The reliable assessment of the integrity of total RNA would need the 3':5' assay.
RNA分离纯化是分子生物学实验的第一步, 也是比较关键的一步。但是多种内外因素制约了RNA的分离纯化, 进而制约了后续实验的开展。RNA固有的化学特性使其容易发生自发降解, RNA的2'位置含有游离的羟基, 在碱性条件下游离的2'-OH会破坏邻近的磷酸二酯键, 从而使RNA链断裂[1], 而且二价金属离子的出现还能加剧这种自发断裂过程[2]。核糖核酸酶(简称RNase)在环境中分布广泛而且非常稳定, 它能抵御100 ℃的高温而且不丧失活性[3, 4], 常规的高温高压蒸气灭菌或使用EDTA都不能使RNase完全失活, 因此, 在实验操作中不慎混入RNase就会不同程度地降解RNA。另外, 内源性的多糖容易与RNA形成难溶的胶状物, 多酚氧化后能与RNA不可逆结合, 它们都制约了RNA分离纯化[5]。系统的对比研究已表明, RNA质量严重影响着荧光定量PCR结果[6, 7]以及DNA芯片的检测准确性[8]。
高质量RNA是后续实验成功的基础与保障, 在RNA抽提后必需对其进行质量检测。因为mRNA只占细胞总RNA的很小一部分且种类复杂, 相反, rRNA占据了绝大部分且种类简单, 因此, 传统上用电泳后的28S rRNA和18S rRNA条带亮度的比值来评估总RNA的完整性(integrity)。18S rRNA、5.8S rRNA和28S rRNA作为整体被转录成多顺反子rRNA前体, 前体进一步加工产生成熟的rRNA[9], 在细胞内18S rRNA的数量与28S rRNA相同。在绝大多数生物中28S rRNA长约4~5 kb, 18S rRNA长约1.9~2.0 kb, 例如人[10]和小鼠[11]的rRNA长度大致相等, 它们的28S rRNA和18S rRNA约为5 kb和2 kb。凝胶电泳后28S rRNA的条带亮度理论上是18S rRNA的2~2.6倍。在实验中, 28S:18S rRNA比值成为了检测总RNA完整性的事实标准, 比值小于2表明降解, 比值大于等于2表明总RNA完整性良好。
在虾蟹类总RNA完整性检测中, 凝胶电泳检测技术也已广泛应用。多位研究者采用不同的方法提取了多种虾蟹类总RNA, 但是电泳之后出现了2种不同的现象。李晓英等[12]和李少菁等[13]分别提取了日本沼虾(Maacrobroachium nipponens)和锯缘青蟹(Scylla serrata)的总RNA, 电泳显示28S rRNA和18S rRNA条带清晰, 比例恰当。李吉涛等[14]和王在照等[15]提取了中国对虾(Fenneropenaeus chinensis)总RNA, 观察到了清晰的28S rRNA和18S rRNA条带, 但18S rRNA条带亮度明显大于28S rRNA的亮度; 周向红等[16]也提取了中国对虾总RNA, 只观察到了18S rRNA条带, 28S rRNA条带几乎不可见。28S rRNA条带的减弱或消失暗示着什么?是总RNA降解, 还是凝胶电泳检测技术不适合虾蟹类总RNA完整性的检测?为探究这一问题, 本文以凡纳滨对虾(Penaeus vannamei)为实验对象, 采用TRIzol法抽提总RNA, 并进行变性琼脂糖凝胶电泳检测, 同时采用实时荧光定量PCR技术直接检测mRNA的完整性。
实验所用凡纳滨对虾共3尾, 取自连云港赣榆的对虾养殖场, 活体解剖后取肝胰脏和肌肉, 然后立刻进行总RNA的分离纯化。
取30 mg新鲜肝胰脏或50 mg新鲜肌肉, 液氮研磨成粉, 加1 mL TRIzol试剂(Invitrogen)并混匀, 室温静置5 min; 加200 μ L氯仿, 混匀后4 ℃ 12 000 r· min-1离心15 min; 小心吸取上层水相并加入等体积异丙醇, 12 000 r· min-1离心10 min; 弃上清后, 向RNA沉淀中加入75%乙醇进行漂洗; 离心后弃上清, RNA沉淀于室温干燥, 最后用适量RNase-free水溶解。
参照DNase I说明书(Fermentas)对总RNA进行处理。按1 U DNase I处理1 μ g总RNA的比例制备反应液, 37 ℃水浴30 min, 最后参照《分子克隆实验指南》(第3版), 按常规苯酚/氯仿抽提方法纯化总RNA。
参照《分子克隆实验指南》(第3版)的方法配制甲醛变性琼脂糖凝胶和MOPS电泳缓冲液。在RNA样品中加入MOPS电泳缓冲液、甲醛和甲酰胺, 先60 ℃保温10 min紧接着冰上速冷以去除RNA二级结构, 取适量混合液电泳, 电泳后拍照。
RT反应按RevertAidTM H Minus First Strand cDNA Synthesis kit说明书(Fermentas)操作, 取2 μ g总RNA, 以Oligo (dT)为引物进行反转录反应, 反应生成第1链cDNA, 10× 稀释后备用。
从GenBank中随机选取2个内参基因ACT(β -actin, AF300705, 长1 320 bp)和eEF1A(elongation factor 1-alpha, GU136229, 长1 547 bp)来设计引物, 所有引物均由生工生物工程(上海)股份有限公司合成。每个内参基因设计3对引物, 其中1对用于内参序列克隆, 另外2对用于定量PCR检测, 2对定量引物分别位于基因序列的5'和3'端。具体的引物序列、退火温度以及扩增属性等见表1。
| 表1 引物序列和扩增属性 Table 1 Primer sequences and amplicon characteristics |
使用克隆引物分别扩增ACT和eEF1A基因序列。2个基因的PCR体系和反应条件均相同, 扩增体系50 μ L, 含有1× PCR Buffer, 2 mmol· L-1 MgCl2, 200 μ mol· L-1 dNTP, 0.4 μ mol· L-1上、下游引物, 2 U Dream Taq DNA聚合酶(Fermentas)以及1 μ L第1链cDNA。反应条件为95 ℃预变性3 min; 95 ℃变性30 s, 56℃退火30 s, 72℃延伸70 s, 30个循环; 72 ℃延伸5 min。扩增反应结束后, 一部分PCR产物用于测序, 另一部分用于切胶回收。切胶回收参照SanPrep柱式DNA胶回收试剂盒(生工)说明书进行, 回收产物稀释1万倍后用作标准品。
使用定量引物分别对ACT和eEF1A基因的5'和3'端进行定量分析, 实时荧光定量PCR反应在Bio-Rad的IQ5定量PCR仪上进行。所有定量PCR反应体系均相同, 都为20 μ L, 其中包含10 μ L 2× SYBR® Premix Ex TaqTM II(TaKaRa)、0.2 μ mol· L-1定量PCR引物和1 μ L第1链cDNA。所有反应条件也相同, 都采用两步PCR法进行扩增, 即95 ℃预变性1 min; 95 ℃变性10 s, 57 ℃延伸30 s, 45个循环; 循环结束后, 从55 ℃缓慢升温至95 ℃, 制备熔解曲线。同时以标准品为模板进行定量PCR, 以校正5'和3'端的定量结果。反应还设有无模板对照(NTC), 每个反应设3个复孔。
将定量PCR仪记录到的所有原始荧光数据从IQ5 Optical System Software v2.1(Bio-Rad)导入到LinRegPCR软件[17], 并由LinRegPCR计算得到平均扩增效率(E)、荧光阈值(threshold)、循环阈值Cq以及初始模板的荧光值N0(N0=threshold /ECq), 最后通过下列公式计算每个基因的3'靶序列与5'端靶序列比值:
3':5' ratio=(N03'/N03'S)/(N05'/N05'S)。
式中, 3':5' ratio表示某个基因的3'端靶序列拷贝数与5'端靶序列拷贝数的比值, N05'表示被检样品5'端靶序列的荧光值, N03'表示被检样品3'端靶序列的荧光值, N05'S表示标准品5'端靶序列的荧光值, N03'S表示标准品3'端靶序列的荧光值。所有数据的计量值采用平均数± 标准差表示。
采用核酸蛋白测定仪(Bio-Rad)分别在230、260和280 nm波长下测量了RNA样品的吸光值。肝胰脏总RNA样品的D260/D230=2.57± 0.22, D260/D280=1.96± 0.05; 肌肉总RNA样品的D260/D230=2.47± 0.13, D260/D280=1.91± 0.09, 表明本研究提取的凡纳滨对虾总RNA样品纯度较高, 其纯度适合进行后续实验。
从肝胰脏和肌肉总RNA样品中都检测出了明亮、清晰的18S rRNA条带, 但是在肌肉样品中检测不到28S rRNA条带, 在肝胰脏RNA样品中仅检测到一条极微弱的28S rRNA条带(图1)。随后用Quantity One软件(Bio-Rad)测量电泳条带的光密度值, 肝胰脏的28S:18S rRNA=0.03± 0.01, 肌肉的28S:18S rRNA=0。
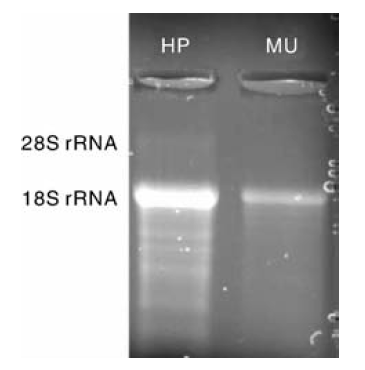 | 图1 总RNA的变性琼脂糖凝胶电泳 HP, 肝胰脏的总RNA; MU, 肌肉的总RNAFig.1 Denaturing agarose gel electrophoresis of total RNA HP, Total RNA extracted from hepatopancreas; MU, Total RNA extracted from muscle |
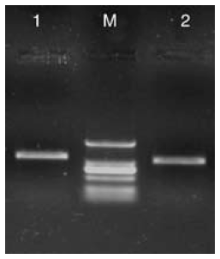
以总RNA为模板, 采用RT-PCR扩增, 得到了ACT和eEF1A基因的特异性扩增产物, 电泳条带约为1 150和1 050 bp, 与设计的扩增产物大小相符(图2), 测序结果也表明, 扩增产物序列分别与ACT和eEF1A基因序列一致。
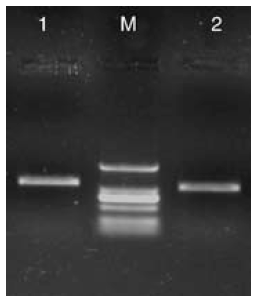 | 图2 扩增产物电泳图 1, ACT基因的扩增产物; 2, eEF1A基因的扩增产物; M, DNA markerFig.2 Electrophoresis of amplicons 1, PCR product of ACT gene; 2, PCR product of eEF1A gene; M, DNA marker |
每对引物的扩增曲线都呈现典型的S型曲线, 基线平整、指数区明显。每对引物的扩增产物在熔解曲线上都只具有特异的单一峰(图3), 无模板对照的扩增曲线呈水平直线状, 说明扩增产物中不存在引物二聚体或非特异性产物, 定量体系的特异性较高、无污染。
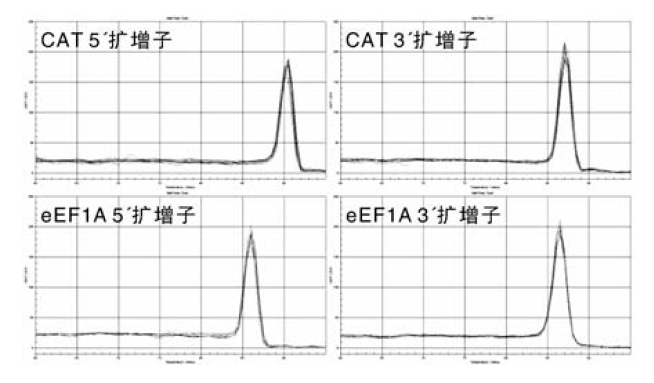 | 图3 定量PCR反应特异性检测Fig.3 Verification of specific products from qPCR reactions |
采用LinRegPCR计算得到的4组扩增的平均扩增效率介于1.881~1.904, 扩增子的平均Cq值介于15.482~17.646(表1), 复孔间Cq值的最大差异为0.399, 最小差异为0.063, 平均差异为0.210± 0.112。表明本实验操作的扩增效率良好、重复性好、结果可靠。
本实验中ACT和eEF1A基因的5'扩增子都远离mRNA的3'末端, 距离3'末端都有1 200 bp以上, ACT和eEF1A基因的5'与3'扩增子的跨度分别为953和800 bp, 它们的3':5' ratio分别为2.79± 0.56和1.53± 0.14。
利用总RNA电泳来评估mRNA完整性的基础假设是样品中rRNA与mRNA具有相同的降解速率, 但是实际上并不清楚rRNA降解与mRNA质量之间的关系[18], 例如健康年老妇女外周血的28S:18S rRNA比值就是小于1[19]。此外, 28S:18S rRNA比值还受到电泳条件、RNA上样量、溴化乙锭荧光的饱和度等多种实验操作的影响, 因此, 凝胶电泳并不是一个可靠的检测技术[18]。
相反, 3':5'分析(3':5' assay)是一项完全不依赖rRNA的直接检测mRNA完整性的技术, 已在基因芯片领域用于检测RNA完整性[20, 21]。3':5'分析技术通过直接检测少数内参基因mRNA的3':5' ratio来检测全体mRNA的完整性, 其基础假设是总RNA样品中所有mRNA具有相同的降解速率。该方法以Oligo (dT)为引物从3'端开始反转录, 若mRNA发生部分断裂, 那么5'端cDNA产物就比3'端的少, 也就是说mRNA完整性越差, 5'端cDNA产物就越少, 因此, 可以通过3':5' ratio来反映mRNA完整性[18, 22]。与凝胶电泳检测技术相比, 3':5'分析技术能更直接、更可靠地检测mRNA完整性。
Nolan等[22]详细描述了基于定量PCR的3':5'分析技术并提出了一个阈值, 即质量良好且能用于后续实验的RNA的3':5' ratio≤ 5。Die等[23]进一步系统地研究了该技术, 指出在3':5'分析中5'扩增子必须远离mRNA的3'末端, 5'扩增子与3'扩增子的间距不同时3':5' ratio的阈值就不同, 5'与3'扩增子的跨度为700 bp时阈值为1.77, 跨度为900 bp时阈值为1.97, 跨度为1 100 bp时阈值为4.43, 大于阈值表示RNA的降解程度不再适合后续的基因表达研究。综合上述观点, 本文提取的凡纳滨对虾mRNA质量良好, 其完整性可用于后续实验。
按照传统观点, 电泳结果(图1)显示本研究提取的凡纳滨对虾总RNA样品已严重降解, 其完整性不适合后续实验。但是我们还是从这种RNA中成功地扩增出了长约1 100 bp的2个基因序列。类似现象在其他虾蟹中也有发生, 虽然抽提的中国对虾总RNA的28S:18S rRNA比值远远小于2, 但是王在照等[15]和周向红等[16]还是分别从中扩增出了697和1 045 bp的基因序列。进一步的3':5'分析更是直接表明本研究提取的凡纳滨对虾mRNA完整性良好。上述所有证据均表明, 凝胶电泳低估了虾蟹类总RNA的完整性, 凝胶电泳技术不适合检测虾蟹类总RNA完整性。
造成凡纳滨对虾28S rRNA条带消失或减少的原因可能与28S rRNA上的隐裂(hidden break)[24]有关。一些原口动物(Protostome, 包括环节动物、软体动物、节肢动物)会在28S rRNA序列的D7a扩展区自发地切除一小段rRNA序列, 从而产生28Sα 和28Sβ 两个rRNA片段[25]。进一步的研究表明这种切割与28S rRNA序列本身有关, Basile-Borgia等[26]用非洲爪蟾(Xenopus laevis)卵母细胞表达异源的尖眼蕈蚊(Sciara coprophila)rDNA基因, 发现尖眼蕈蚊28S rRNA仍然发生了断裂。但是目前尚未发现任何参与隐裂位点识别与切割反应的保守序列, 隐裂的机制知之甚少[25]。在细胞内28Sα 和28Sβ rRNA片段在断裂处通过广泛的氢键结合在一起[27], 遇到变性条件, 28Sα 和28Sβ rRNA片段就会彼此分离[25], 因此, 变性电泳时通常看不见完整的28S rRNA条带。
在实际操作过程中, 我们还发现28S rRNA条带消失或减少的现象在剖刀蛏(Cultellus scalpellum)和菲律宾蛤仔(Ruditapes philippinarum)等软体动物中也发生, 推测其可能也与隐裂有关。如何正确地评估这些水产无脊椎动物总RNA完整性成为实践上急需解决的问题。我们认为, 首先用凝胶电泳对其进行快速和初步检测, 以电泳出现清晰、无弥散、无拖尾的18S rRNA条带为标准, 然后利用3':5'分析技术对其进行细致的检测。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|