
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
鸡卵清蛋白基因调控序列的克隆与载体构建
[黄菁 , 朱志伟, 陈晓宇, 于福先, 潘建治
, 朱志伟, 陈晓宇, 于福先, 潘建治* ]
, 朱志伟, 陈晓宇, 于福先, 潘建治|
|


作者简介:黄菁(1983—),女,浙江舟山人,博士,助理研究员,从事动物生物工程研究。E-mail:xiaojingyu1102@gmail.com
卵清蛋白(ovalbumin)基因在鸡基因组中只有一对等位基因,却能每天合成分泌多达2 g的蛋白,占据卵白蛋白质的50%以上,是外源基因载体表达调控构件的首选。该研究从输卵管特异性启动子方面着手,通过对卵清蛋白基因启动子的筛选优化,找出启动子增强因子的位置以及组织特异性区域。将卵清蛋白基因上游-922~-2 073和-2 801~-3 100区域平均分成12个长度约为150 bp的序列,分别插入到-921~+38序列的上游,成功构建12个系列表达载体,为进一步筛选短缩版优化启动子提供材料;卵清蛋白基因第一内含子区域截断成300 bp左右的迷你内含子序列,成功构建8个迷你内含子系列载体,为筛选优化的迷你内含子提供必要的材料;成功分离鸡输卵管上皮细胞并优化电转染条件,通过荧光素酶活性检测初步筛选出具有最强活性重组质粒pGL4-UP-1412和内含子重组质粒pGL4-mini-intron-3,同时推断出若干包含增强子序列区域。
There was only one allele of ovalbumin gene in the chicken genome, but it could synthesize and secrete 2 g protein per day, which were accounting for more than 50% of the albumin protein and became the preferred choice in the regulation of exogenous gene expression. This study aimed to identify promoter enhancer and tissue-specific regional location factor by screening the optimization of ovalbumin gene promoter. The upstream -922~-2 073 and -2 801~-3 100 of ovalbumin promoter were divided into 12 regional which average sequence length were about 150 bp, and inserted into the upper reaches of -921~+38 sequences, those 12 series successfully constructed expression vectors provided materials for further optimization promoter with a shortened version. The first intron region of ovalbumin promoter was truncated around 300 bp of mini intron sequences, and successfully constructed 8 mini intron series of vectors. We also successfully separated chicken oviduct epithelial cells and optimized the electricity transfection conditions. In this study, the promoter region with strongest activity pGL4-UP-1412 and pGL4-mini-intron-3 were screened through detected luciferase activity of the initial screening recombinant plasmid and intron recombinant, while inferred several regions containing enhancer sequence.
卵清蛋白(Ovalbumin)在鸡基因组中只有一对等位基因, 却能每天合成分泌多达2 g的蛋白, 占据卵白蛋白质的50%以上, 自然成为外源基因载体表达调控构件的首选。至今, 以家禽输卵管定位表达为目标的基因导入研究, 绝大部分采用了卵清蛋白基因的调控序列。 然而, 即便采用了卵清蛋白的调控序列, 也不能高效诱导出外源蛋白的大量合成和分泌。迄今, 在美日韩等国制备的生物反应器转基因家禽卵中, 外源蛋白占卵白蛋白的极少超过1‰ 。Lillico等 [1]的研究中采用了卵清蛋白基因上游2.8 kb的片段, 实现了迄今最成功的输卵管特异表达, 卵白中人干扰素含量达到平均38 mg· L-1, 最高的个体也仅为426 mg· L-1。相比之哺乳动物细胞培养体系的水平(10 g· L-1左右)仍然存在相当差距。Kwon等[2]进一步缩短启动子长度至1.35 kb用于转基因鹌鹑, 虽然保持了输卵管特异性, 但在卵白中表达重组人白细胞介素-1受体拮抗蛋白(rhIL1RN)的含量更低, 仅为88.7~233.8 μ g· L-1。Zhu等[3]采用卵清蛋白基因5'端上游长达7.5 kb和15 kb的调控序列制作嵌合体鸡表达单克隆抗体, 虽然表达量较高(750 mg· L-1), 但输卵管特异性不强, 也超过了病毒载体允许插入的长度。
外源基因表达量的高低, 除受到基因组插入位置效应等随机因素的影响外, 主要与转基因载体中启动子等元件的活性有关, 单纯截取卵清蛋白基因上游的调控序列片段, 难以获得根本性的改善。而利用基因敲入的方法向卵清蛋白基因座定点导入外源基因, 或者将外源基因插入BAC克隆等包含卵清蛋白基因的长片段中, 再导入到基因组, 虽然有可能使远距离存在的潜在调节元件发挥作用, 获得高效率特异性表达, 但受到目前家禽转基因技术的限制, 尚难于在个体水平上付诸实施, 无法获得实验验证。
因此, 在转基因载体的有限长度内, 对表达调控元件, 特别是启动子领域进行优化, 是现实有效的途径, 也是国际性的研究热点。本研究从输卵管特异性启动子方面着手, 通过对卵清蛋白基因启动子的筛选优化, 找出启动子增强因子的位置以及组织特异性区域, 为下一步对增强区域和特异性区域的整合, 制备出具有表达活性高、组织特异性强的优化型启动子提供基础材料。
PCR仪(ABI 9902, 美国应用生物系统公司); 高速冷冻离心机(HITACHI CR21GIII, 日本株式会社日立制作所); 酶标仪(Molecular Devices SpectraMax M5, 美国); 倒置显微镜(Nikon ELIPSE Ti-U, 日本尼康公司); 电转染仪(BTX ECM830, 美国哈佛仪器旗下BTX公司); 内切酶(TAKARA公司); DNA回收试剂盒[TIANGEN, 天根生化科技(北京)有限公司]; 荧光素酶活性检测系统(ONE-Glo Luciferase Assay system, Promega公司)。
1.2.1 鸡卵清蛋白基因上游调控序列的克隆
参考GenBank中鸡卵清蛋白基因组序列(NC_006089), 利用生物软件Primer Premier 5和DNAMAN设计添加了酶切位点的引物, PCR扩增系列目的片段。引物名称和序列详见表1。
| 表1 引物序列及酶切位点 Table 1 Sequences of primers and their restriction sites |
将转录起始位点作为+1位置, 本试验克隆的序列按照其长度分别命名详见表2。
以实验室已有的pGL4-921作为模板, TransTaq High Fidelity(HiFi)PCR SuerMix II PCR Kit扩增目的片段X— Y。用引物(F-3100, R-2981), (F-3010, R-2891), (F-2920, R-2801), (F-2703, R-1916), (F-1937, R-1776), (F-1803, R-1636), (F-1659, R-1522), (F-1547, R-1387), (F-1412, R-1251), (F-1275, R-1122), (F-1148, R-1023), (F-1048, R-922)进行PCR扩增反应。PCR产物在2%琼脂糖凝胶中进行电泳检测, 用DNA纯化回收试剂盒(离心柱型, TIANGEN)进行割胶回收。
| 表2 序列名称及序列位置 Table 2 Name and position of sequences |
1.2.2 鸡卵清蛋白基因上游调控序列变异荧光素酶报告基因载体构建
将回收得到的上游DNA片段进行Kpn Ⅰ 酶切, 酶切后产物用DNA纯化回收试剂盒(离心柱型, TIANGEN)进行纯化回收。将酶切后回收得到的UPs目的片段与pGL4-921双酶切产物按照比例进行连接, 然后转化大肠埃希菌。采用菌液PCR法筛选出阳性菌落。挑选正向连接检测为阳性的菌液送生工生物工程(上海)股份有限公司测序。
1.3.1 卵清蛋白基因第一内含子全序列及系列分割序列克隆
参考GenBank中鸡卵清蛋白基因组序列(NC_006089), 利用生物软件Primer Premier 5和DNAMAN设计8对添加了酶切位点的引物, PCR扩增系列包含或不包含内含子的目的片段。具体引物序列详见表3。方法同上。
| 表3 PCR引物序列及酶切位点 Table 3 Sequence of primers and their restriction sites |
1.3.2 迷你内含子系列载体构建
以纯化回收得到的OV基因为模版, PCR扩增目的片段OV+38/OV+1640/OV-合成, 胶回收法纯化回收PCR产物。方法同上。
将OV+38/OV+1640/OV-合成的PCR产物进行Kpn Ⅰ , Xho Ⅰ 双酶切, 同时将质粒pGL4.10 Vector进行Kpn Ⅰ , Xho Ⅰ 双酶切。37 ℃酶切2 h后, 将每个酶切产物进行切胶回收、连接、转化, 挑选阳性克隆子测序鉴定。
1.4.1 鸡输卵管上皮细胞分离培养与转染
输卵管上皮细胞的分离培养步骤如下:
(1)放血处死试验鸡, 尽量将血放干净, 体内如果瘀血太多, 输卵管上也会存在大量血丝, 分离上皮细胞时会有较多的红细胞掺杂进来。
(2)将处死的鸡置于托盘中, 腹部朝上, 去除腹部及鸡右侧腿部羽毛(去除之前可喷洒医用乙醇, 避免鸡毛散飞), 用灭菌剪刀剪开腹部及鸡右侧皮肤, 换剪刀镊子, 小心剪开腹部肌肉, 去除脂肪, 取出输卵管膨大部, 置于超净台内准备好的PBS中。
(3)选择较好的膨大部3~5 cm, 用小剪刀、小镊子小心去除膨大部外层的膜和血管。输卵管内层为褶皱状, 去除外层系膜有利于褶皱部展开, 消化时与胰酶充分接触。此外, 毛细血管一般分布在系膜与组织之间, 去除系膜有利于去除血管, 减少血细胞的污染。
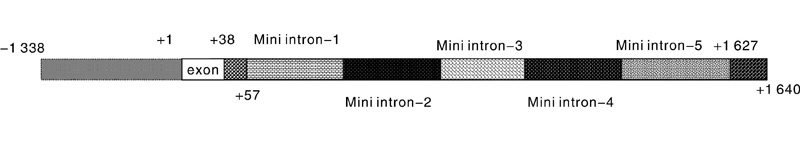 | 图1 第一内含子分区示意图Fig.1 The partition schematic of the first intron |
(4)将血管、系膜去除干净的输卵管部分翻折, 内层朝外。利用小剪刀的把手进行刮除附着的黏液成分, 在PBS中进行彻底的清洗。(去除膜以后的输卵管组织特别薄, 不能用尖锐利器进行刮除)。
(5)将清洗干净的组织整块至于适量0.2%胶原酶中, 水浴锅中预热5 min, 然后于37 ℃摇床85 r· min-1消化5~8 min, 250 g离心4 min, 去除上清, PBS清洗2遍, 离心去上清。
(6)加入3倍体积的胰酶37 ℃摇床85 r· min-1消化10~15 min, 超净台内吹打数次, 取少量消化液显微镜下观察细胞数量。若细胞数较少, 可适当延长消化时间, 若能够观察到大量密布细胞时即可终止消化。
(7)将吹打数次的消化液于200目筛过滤, 将滤液收集入新的50 mL离心管中, 加入适量血清终止胰酶的消化, 250 g离心4 min, 去除上清。PBS清洗2次, 离心去上清。
(8)加入适量PBS进行洗涤, 250 g离心4 min后去除上清, 加入适量含10%胎牛血清、1% 双抗的DMEM重悬, 混匀后置于10 cm培养板中, 37 ℃, 5%二氧化碳培养, 24 h后显微镜下观察细胞贴壁情况。
细胞转染参照lipofecatmine 2000(Thermo Fisher)试剂使用说明书。
1.4.2 荧光素酶活性测定
利用ONE-Glo Luciferase Assay system(Promega)对不同重组表达载体在细胞中的荧光素酶表达进行测定。试验步骤如下:
(1)提前将光栅式多功能酶标仪开启, 设定化学发光测定程序。
(2)准备进行荧光素酶活性测定的细胞去除培养液, PBS清洗2~3遍。
(3)向每个孔内加入100 μ L PBS+150 μ L ONE-Glo反应液, 避光, 室温条件下轻轻晃动培养板将细胞充分裂解(至少3 min)。
(4)将24孔板中的细胞裂解液吹打数次, 每孔取200 μ L裂解液吸取至白色96孔酶标板。
(5)将加入裂解液的96孔酶标板放入多功能酶标仪, 测定荧光素酶活性。
2.1.1 pGL4-UPS表达载体菌液PCR阳性结果
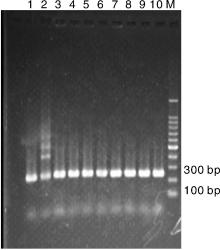
上游调控序列各片段PCR产物回收之后, 12个片段均介于120~200 bp之间, 符合预期大小。所有重组质粒的菌液PCR检测以pGL4.10载体上的pGL3+(TAGCAAAATAGGCTGTCCC)为上游引物, OVsR为下游引物, 12个重组载体中的目的片段大小存在一定差异, 所以经PCR扩增后, 正确的连接将会分别扩增出介于200~300 bp之间, 约为220 bp左右的片段。图2显示为其中一个表达载体PGL4-UP-2073菌液PCR呈阳性, 所有测序验证结果表明该载体构建成功。
2.1.2 pGL4-UPS表达载体构建的酶切鉴定成功
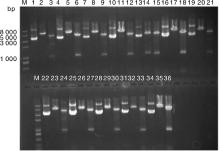
菌液鉴定为阳性的质粒经过测序验证, 选取测序无突变克隆子提取质粒, Kpn Ⅰ , EcoR Ⅴ 双酶切鉴定。所有质粒总长度均为5 000 bp左右, 经Kpn I, EcoR Ⅴ 双酶切后, 将质粒切成长度分别为4 020, 921和120~160 bp的3部分。由图3可见, 所有质粒的酶切检测均获得预期片段, 质粒构建成功。泳道1, 4, 7, 10, 13, 16, 19, 22, 25, 28, 31, 34为构建的12个重组质粒。每个质粒后的2个泳道分别为相应质粒的限制性内切酶EcoR Ⅴ 的单酶切和Kpn Ⅰ , EcoR Ⅴ 双酶切电泳图。
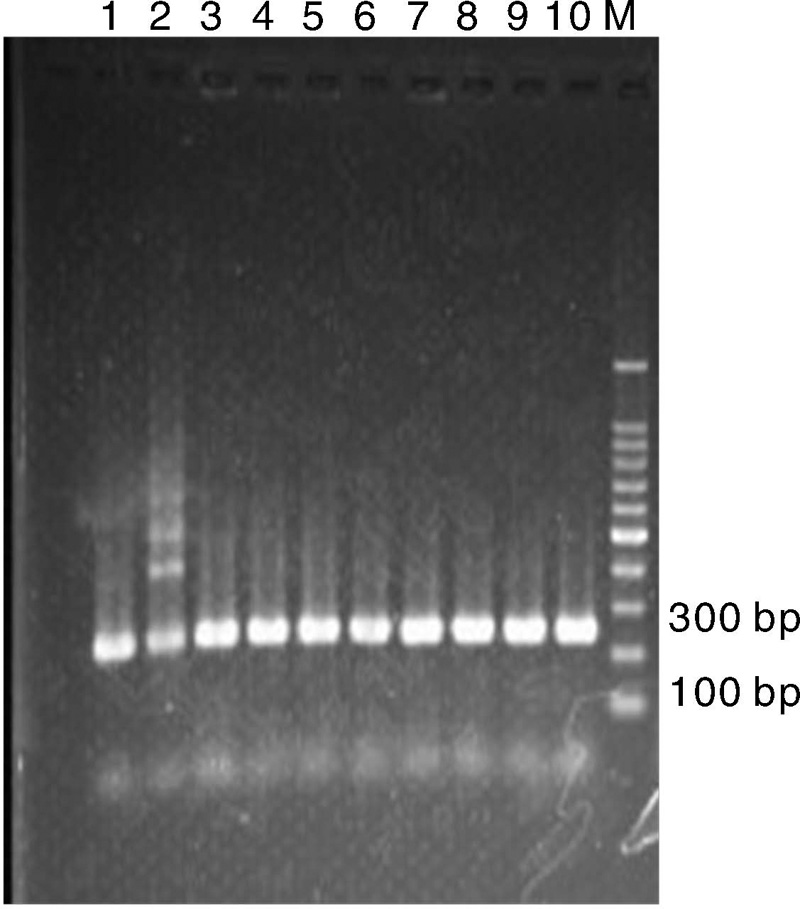 | 图2 pGL4-UP-2073正连接菌液PCR检测结果Fig.2 PCR result of the positive ligation of pGL4-UP-2073 plasmid |
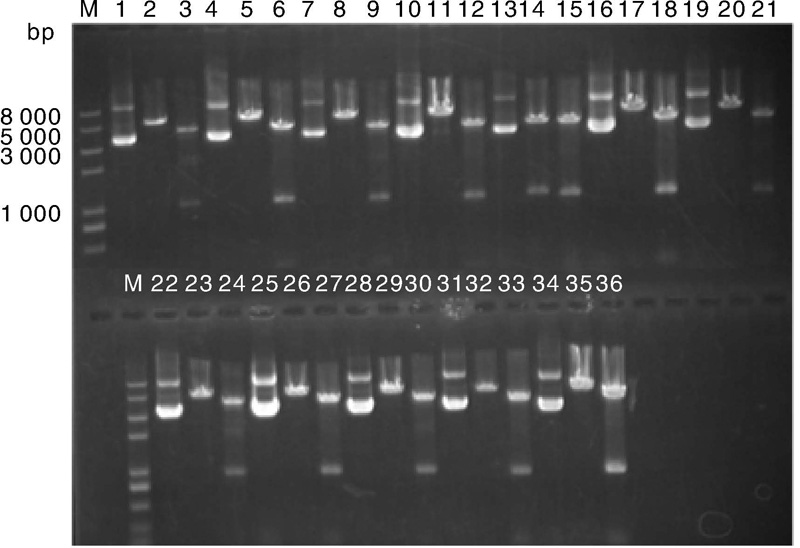 | 图3 pGL4-UPs质粒酶切鉴定结果 泳道1, 4, 7, 10, 13, 16, 19, 22, 25, 28, 31, 34为构建的12个重组质粒; 泳道2, 5, 8, 11, 14, 17, 20, 23, 26, 29, 32, 35为各质粒经由EcoR Ⅴ 的单酶切; 泳道3, 6, 9, 12, 15, 18, 21, 24, 27, 30, 33, 36为各质粒经由Kpn Ⅰ , EcoR Ⅴ 双酶切。Fig.3 Results of restriction enzyme digestion for pGL4-UPs plasmids |
2.2.1 卵清蛋白基因第一内含子序列成功克隆
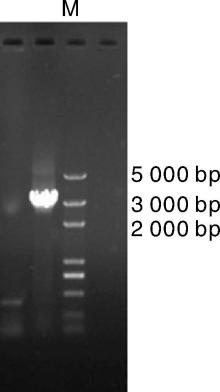
利用PCR方法克隆鸡卵清蛋白基因第一内含子-1 338~+1 798位置的目的片段, 纯化回收后电泳图。由图4可见, 目的基因长度为3 136 bp, 目的条带的位置符合预期, 克隆成功。
2.2.2 基础载体及迷你内含子系列载体成功构建
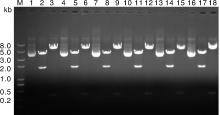
经过菌液PCR阳性筛选和测序验证之后, 将无序列突变质粒进行酶切鉴定, 如图5所示, 所有质粒的酶切检测均获得预期片段, 质粒构建成功。泳道1, 4, 7, 10, 13, 16为构建成功的质粒, 每个质粒后的2个泳道分别为相应质粒的限制性内切酶EcoR Ⅴ 单酶切和Kpn Ⅰ , EcoR Ⅴ 双酶切电泳图。
2.3.1 鸡输卵管上皮细胞培养状态良好
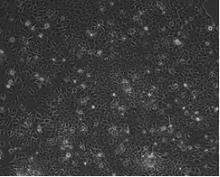
由图6可见, 本试验成功地从产蛋母鸡的输卵管分离到上皮细胞, 呈现出良好的上皮细胞形态, 成纤维细胞较少。
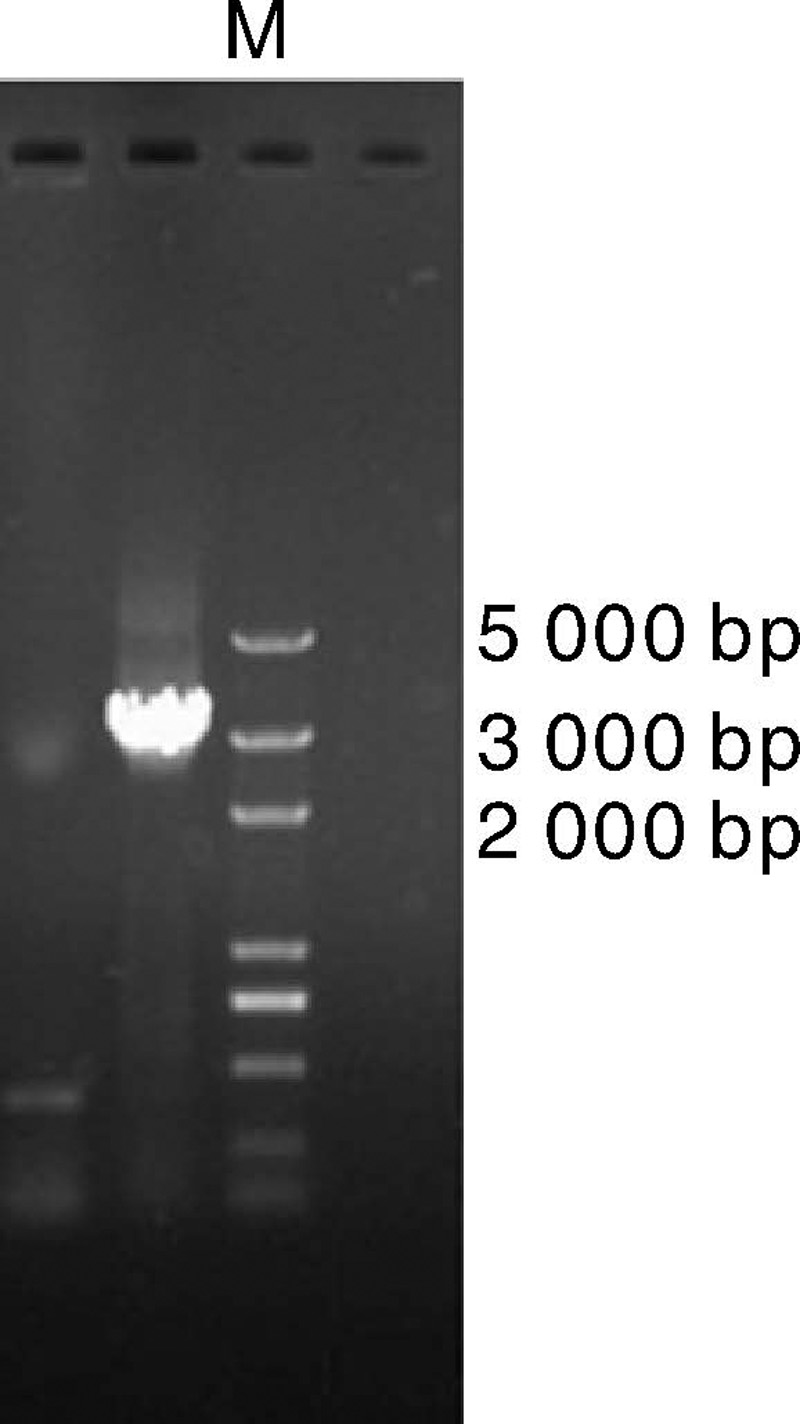 | 图4 第一内含子序列克隆回收后凝胶电泳图Fig.4 Gel electrophoresis of the first-intron sequence after cloning and purification |
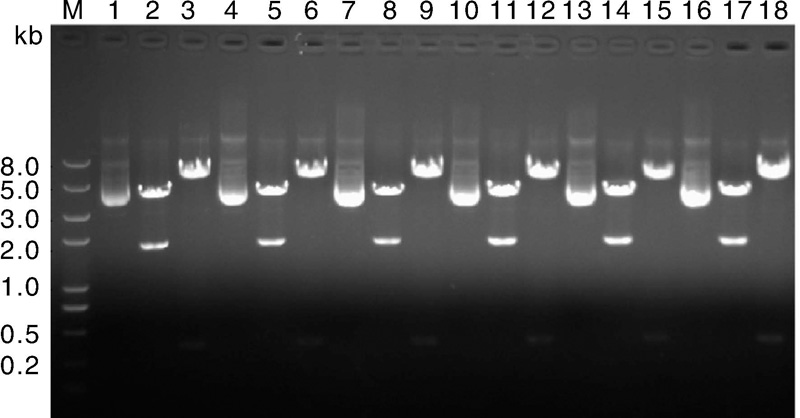 | 图5 pGL4-mini-introns质粒酶切鉴定结果 泳道1, 4, 7, 10, 13, 16为构建成功的质粒; 泳道2, 5, 8, 11, 14, 17为各质粒经由EcoRV单酶切; 泳道3, 6, 9, 12, 15, 18为个质粒经由KpnI, EcoRV双酶切。Fig.5 Results of restriction enzyme digestion for pGL4-mini-introns plasmids |
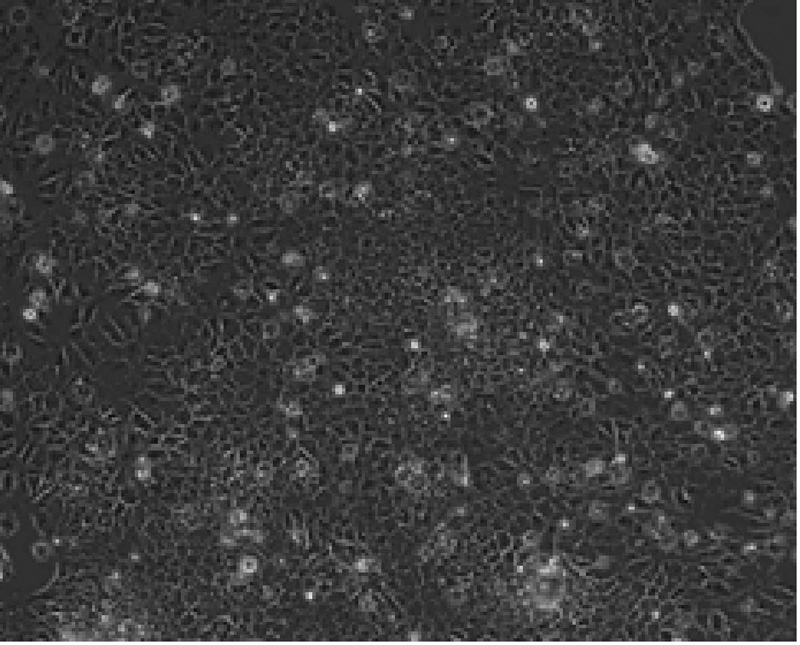 | 图6 鸡输卵管上皮细胞形态Fig.6 Cell morphology of chicken oviduct epithelial cell |
2.3.2 确定最优转染条件
根据有关文献, 将电转染电压初步设定为250 V, 利用293T细胞进行电转染时间的优化, 将电击时间设1, 3, 5, 7, 9, 11 ms等6个实验组, 结果显示在同等电压下, 7 ms以上细胞死亡率达30%以上, 1 ms时, 转染效率很低, 在保证细胞成活率的前提下, 选择5 ms作为最佳电击时间。根据此结果, 在输卵管上皮细胞上进行电转染条件的进一步优化。以250 V, 5 ms为基础, 固定电击时间, 设150, 200, 250, 300, 350 V等5个实验组进行探究。实验结果显示:300 V, 5 ms条件时, 相较于低电压电转条件, 死亡细胞数稍多, 悬浮于培养液中, 转染效率高, 可达20%左右, 如图7所示, 而电压为350 V时细胞死亡数过多, 不利于后续试验, 因此确定最佳电转染条件为300 V, 5 ms。
 | 图7 鸡输卵管上皮细胞电转染效果Fig.7 Electric transfection efficiency of chicken oviduct epithelial cell |
2.3.3 卵清蛋白基因上游调控序列表达活性
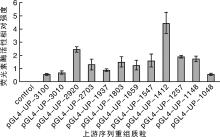
荧光素酶检测结果显示, 重组质粒pGL4-UP-1412表达活性最强(图8), 表明在鸡卵清蛋白基因上游-1 412~-1 251 bp序列部分可能存在有效调控元件。
2.3.4 迷你内含子系列载体表达活性
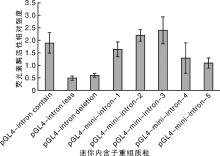
荧光素酶检测结果显示, 内含子重组质粒pGL4-mini-intron-3表达活性最强(图9), 说明在内含子+678~+1 008位置存在对转录活性有增强作用的区域。整体比较pGL4-mini-intron-1, pGL4-mini-intron-2, pGL4-mini-intron-3, pGL4-mini- intron-4和pGL4-mini-intron-5, 结果显示, 表达活性呈先降后升的趋势, 表明在+1 298~+1 628区域可能存在表达抑制性因子。同时推断出若干包含增强子序列区域。
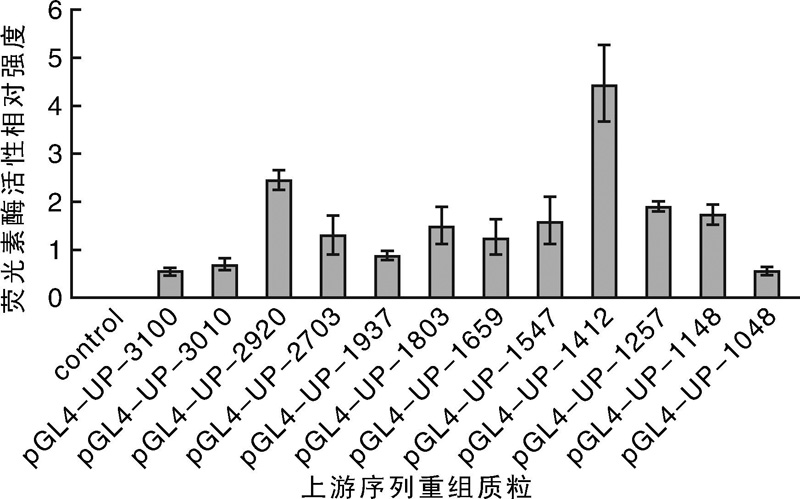 | 图8 pGL4-Ups质粒荧光素酶活性检测结果Fig.8 Luciferase activity of pGL4-UPs plasmids |
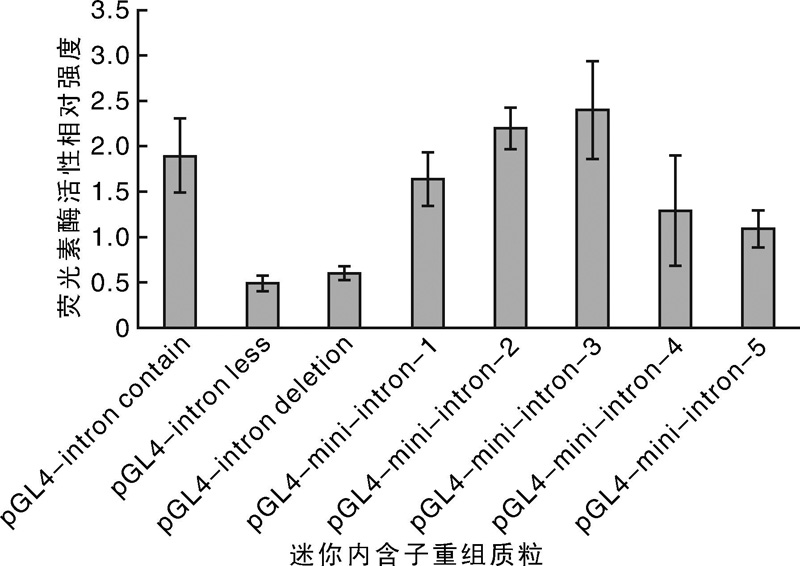 | 图9 pGL4-mini intron质粒荧光素酶活性检测结果Fig.9 Luciferase activity of pGL4-mini intron plasmids |
与哺乳动物相比, 禽蛋作为表达重组蛋白的载体, 具有多方面显著优势:家禽繁殖周期短, 能在较短周期内建群; 家禽饲养成本低、产蛋量高, 一段湿质量50 g的产卵期输卵管膨大部每天能产生4 g蛋白, 蛋白合成分泌的效率惊人; 卵白中的蛋白不会漏入血液循环, 危害动物本身的健康; 禽蛋蛋白成分简单, 具有天然无菌环境, 稳定性好, 便于提取纯化; 输卵管细胞能表达糖蛋白和磷蛋白等复杂结构的蛋白质, 结构较牛羊乳腺更接近于人[4]; SPF饲养技术成熟, 产品安全性高等。以上特点使家禽被视为生产重组蛋白的理想生物反应器体系, 其生产成本有可能降至细胞培养法的1%以下, 也大大低于转基因牛羊。蛋白中的蛋白质稳定, 药物蛋白在鸡蛋中有比较长的半衰期, 因此禽类输卵管生物反应器以其卓越的优势必将成为生物科学领域研究的热点之一, 必将成为研究和投资的重点, 成为医疗性蛋白生产的理想途径。
商业化的载体所容纳外源基因具有一定的长度限制, 因此, 在表达载体的有限长度内, 对表达调控元件进行优化, 是一种现实有效的途径, 也是国际性的研究热点。此外, 外源基因的表达量高低, 除受到基因组插入位置效应的影响外, 主要与转基因在体内启动子等元件的活性有关, 单纯截取卵清蛋白基因上游的调控序列片段, 难以获得根本性的改善。在本试验结果的基础上, 制备出具有表达活性高、组织特异性强的优化型启动子, 调控蛋白基因的高效表达, 从而解决载体容纳量低和外源基因在体内表达量低的问题。
生物反应器可以生产种类繁多的有用蛋白, 包括单克隆抗体、疫苗、激素、生长因子、酶、血清蛋白等, 这些大多难于从自然界原料中分离提取, 或者自然界根本不存在。微生物(大肠埃希菌和酵母)发酵表达系统是目前最为常见的基因工程蛋白生产手段, 但对于众多具有复杂结构的蛋白而言, 难于从微生物获得正确表达, 立体构造和翻译后修饰状态的不同, 影响到蛋白质的功能。因此, 需要借助高等动物细胞培养系统或动物个体来进行重组蛋白生产。
本研究成功获得pGL4-UP-2703, pGL4-UP-1937, pGL4-UP-1803, pGL4-UP-1659, pGL4-UP-1547, pGL4-UP-1412, pGL4-UP-1257, pGL4-UP-1148, pGL4-UP-1048, pGL4-UP-3100, pGL4-UP-3010, pGL4-UP-2920等12种鸡卵清蛋白上游调控序列重组报告基因载体; 及pGL4-intron-contain, pGL4-intron-less, pGL4-intron-delation, pGL4-mini-intron-57, pGL4- mini-intron-368, pGL4- mini-intron-678, pGL4- mini-intron-988, pGL4- mini-intron-198等8种鸡卵清蛋白第一内含子迷你化重组报告基因载体; 成功分离培养了鸡输卵管原代上皮细胞, 并优化电转染条件, 为后期实验研究提供了有用的实验数据; 所构建的载体均具有调控荧光素酶基因表达的活性。通过比较UPs等20种序列的启动子活性, 为找出存在于鸡卵清蛋白基因上游调控序列中及第一内含子中的增强子、抑制子等功能性区域提供了新的数据, 但要制作出高活性、高特异性的启动子还须进一步探索研究。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|