
{kind=link}
{kind=link}
{kind=link}
稻瘟病菌分泌蛋白组学研究
[崔书建1  , 王教瑜
, 王教瑜2 , 姜华2 , 梁建生1, * ]
, 王教瑜]
|
|


作者简介:崔书建(1980—),男,江苏扬州人,博士研究生,主要从事蛋白质组学研究 。E-mail: cuisj80@aliyun.com
稻瘟病菌分泌蛋白在稻瘟病菌入侵水稻过程中发挥重要作用。收集稻瘟病菌(Guy11菌株)液体培养的上清液,富集蛋白,经过烷基、还原处理和酶解后,采用液相色谱-串联质谱(LC-MS/MS)对酶解肽段进行分离及鉴定,利用信号肽预测工具SignalP和蛋白质亚细胞定位软件Protcomp对鉴定到的蛋白进行生物信息学预测验证。结果显示,共鉴定出30个稻瘟病菌的分泌蛋白,其中:24个蛋白具有N端信号肽,26个蛋白可分泌到胞外,3个蛋白定位在细胞膜上。对其功能进行分类,发现多数为酶类,参与水解及能量代谢。这些酶类和蛋白的发现与进一步研究,将为揭示稻瘟病菌侵染的机理提供帮助。
Secreted proteins of Magnaporthe oryzae play an important role during infection process on rice. In this study, the proteins enriched from the supernatant of the M. oryzae(Guy11 strain) culture were digested after alkylation and reduction treatment. The digested peptides were separated and identified using liquid chromatograph mass spectrometer (LC-MS/MS). Bioinformatic predictions of intracellular localization and function of these identified proteins were carried out using the SignalP and Protcomp softwares. Thirty secreted proteins of Magnaporthe oryzae were identified, among which, 24 proteins contained the signal peptide at the N-terminal, 26 proteins were capable of secreting out of the cell, and 3 proteins were localized on the cell membrane. Functional prediction indicated that most proteins were enzymes involved in hydrolysis and energy metabolism. Identification and further investigation of these enzymes and proteins would contribute to reveal the molecular pathogenesis of Magnaporthe oryzae infection.
稻瘟病菌(Magnaporthe oryzae)能够在水稻生长过程中对其进行全面侵染, 引发稻瘟病[1]。全球范围内各稻区均可发生稻瘟病, 同时, 全球约一半的人口以稻米为主食; 因此, 稻瘟病直接威胁全球粮食供应与粮食安全[1, 2, 3, 4, 5]。稻瘟病菌是研究真菌遗传, 以及植物与病原微生物相互作用的重要模式生物[6]。2005年, 美国科学家完成了稻瘟病菌的全基因组测序, 并公布其基因组草图[2, 3, 4], 有力促进了对稻瘟病菌, 及植物和稻瘟病菌互作分子机制的深入研究。
分泌蛋白是分泌到细胞膜外的蛋白质, 在动植物中均普遍存在, 具有免疫反应、组织器官形态的维持信号传递等功能。在稻瘟病菌侵染植物的过程中, 分泌蛋白也参与其中, 并发挥重要作用。有研究显示, 稻瘟病菌附着胞特异表达的GAS1和GAS2基因均编码分泌蛋白。这2个基因的缺失, 不仅会降低稻瘟病菌附着胞的形成能力, 而且还会降低稻瘟病菌的侵染力[7]。胞外几丁质结合蛋白CBP1研究显示, 该分泌蛋白起着信号传导作用, 帮助稻瘟病菌感知外界环境[8]。大量研究表明, 寄主水稻与稻瘟病菌间存在特异性的识别关系, 即“ 基因对基因” 假说。在水稻中, 每一个显性的抗性基因在稻瘟病菌中都存在一个与之对应的显性的无毒基因[9]。照假说设定的互作体系, 水稻抗病基因的产物会与稻瘟病菌无毒基因的产物相互识别, 开启水稻的防御反应, 从而对稻瘟病菌产生抗性。水稻中有相应的抗病基因存在, 同时稻瘟病菌中又有相应的无毒基因存在, 水稻才可以表现出相应的抗病性[10]。根据无毒基因与抗性基因的“ 基因对基因” 关系, 研究人员现已成功克隆出9个稻瘟病菌无毒基因[11]。这些无毒基因可以分为2类:品种特异抗性的无毒基因, 寄主和非寄主特异性抗性的无毒基因。第一类无毒基因数量最多, 主要包括Avr1-CO39、Avr-Pita、ACE1、AvrPiz-t、Avr-Pia、Avr-Pii和Avr-Pik/km/kp; 第二类基因编码PWL激发子。PWL为一个基因家族, 共计包含4个基因(PWL1、PWL2、PWL3和PWL4), 目前了解相应功能的是PWL1和PWL2[12, 13]。无毒基因的成功分离, 不仅为“ 基因对基因” 学说提供了支持, 也为稻瘟菌致病性的研究提供了材料。无毒基因与寄主互作的特性决定了无毒基因多数为分泌蛋白, 能够从稻瘟病菌细胞分泌到寄主细胞内。稻瘟病菌无毒基因与水稻抗性基因的“ 基因对基因” 关系, 以及无毒基因大多数为分泌蛋白的事实, 使得从分泌蛋白入手分离更多的效应子成为可能, 为进一步探求互作的分子机理提供了手段。
分泌蛋白组一词, 在研究枯草杆菌分泌蛋白时首次提出[14]。含有信号肽的蛋白, 并且能通过内质网-高尔基体途径分泌, 所有这些蛋白组的集合为分泌蛋白组。这些蛋白也被称为经典分泌蛋白[15]。本研究通过液相色谱-串联质谱(LC-MS/MS)对稻瘟病菌的分泌蛋白组进行鉴定和研究, 共鉴定出30个蛋白, 经过生物信息学分析验证, 其中, 24个蛋白含N端信号肽, 26个蛋白可分泌到胞外, 3个蛋白定位在细胞膜上, 只有1个定位在细胞质内。这些蛋白多为酶类, 参与水解及能量代谢, 研究结果可为进一步分离效应蛋白、揭示稻瘟病菌致病机理提供借鉴。
供试菌株Guy11为稻瘟病菌标准菌株, 蒙浙江大学林福呈教授惠赠。
固体完全培养基(CM):将1 g酵母提取物、1 g酪蛋白氨基酸、10 g葡萄糖、2 g蛋白胨、3 g硝酸盐、0.003 g维生素、0.01 g微量元素、15 g琼脂粉, 溶于1 L水中, 灭菌。
稻谷粉培养基(固体):将20 g稻谷粉、1.5 g酵母提取物、15 g琼脂溶于1 L水中, 灭菌。
YEG培养基(液体):将2 g酵母提取物、10 g葡萄糖、3 g KNO3、2 g KH2PO4溶于1 L水中, 灭菌。
1.3.1 菌株活化
菌株活化参照Dioh等[16]的方法。将含有真菌菌株的滤纸片接种于稻谷粉培养基上, 在12 h光照/黑暗交替条件下26 ℃进行活化。培养3 d后, 取一小块边缘新鲜的菌丝接种于CM固体培养基上, 同上述条件继续培养。3 d后, 取1 mm含边缘新鲜菌丝的菌落块放入24孔板中, 每孔加1.5 mL YEG液体培养基, 黑暗条件下培养3 d。
1.3.2 稻瘟病菌分泌蛋白提取及定量
收集YEG培养液, 12 000 r· min-1、4 ℃离心取上清, 3 ku超滤柱去盐富集浓缩, 富集后的样本基于二喹啉甲酸(BCA)比色法蛋白定量试剂盒测定蛋白浓度。
1.3.3 蛋白质酶解
将100 μ g蛋白加入10 mmol· L-1二硫苏糖醇(DTT)中, 56 ℃温浴1 h, 室温下经50 mmol· L-1碘乙酰胺烷基化45 min, 加入2 μ g修饰级别的胰酶、50 mmol碳酸氢铵, pH大于8.0, 37 ℃酶解过夜, 0.1%三氟乙酸(TFA)终止酶反应; 固相萃取小柱(Waters)去盐, 真空浓缩仪抽干, -80 ℃保存。
1.3.4 肽段在线分离及质谱采集
肽段复合物经20 μ L 2%乙腈(CAN)/0.1%甲酸(FA)溶解, 12 000 r· min-1离心10 min, 吸取上清上样, 经反相色谱柱(75 μ m× 15cm, C18, Waters)在线分离。流动相A为2% CAN/0.1% FA, 流动相B为98% CAN/0.1% FA。洗脱液流速设定为300 nL· min-1。液相色谱线性梯度洗脱程序如下:0— 3 min, 97% A, 3% B; 3— 60 min, 97%~75% A, 3%~25% B; 60— 85 min, 75%~52% A, 25%~48% B; 85— 86 min, 52%~20% A, 48%~80% B; 86— 90 min, 20% A, 80% B; 90— 91 min, 20%~97% A, 80%~3% B; 91— 101 min, 97% A, 3% B。分离的肽段经离子源离子化进入质谱(TripleTOF 5600)采集, 采集条件如下:离子源电场电压2.3 kV, 雾化气为4, 气帘为35, 加热温度150 ℃, 图谱采集模式为数据依赖型采集(DDA)模式。一级扫描参数:质荷比(m/z)350~1 300, 累积时间0.25 s。串级扫描参数:采集数目为25, m/z 100~1 500, 累积时间0.1 s, 动态排除时间25 s, 碰撞能量波动状态为激活状态。
1.3.5 数据库搜索及生物信息学预测分析
蛋白质鉴定分析采用质谱结果分析软件Protein Pilot 4.5进行, 数据库下载于美国国立生物技术信息中心(NCBI)网站, 以“ Magnaporthe oryzae” 为关键词条, 共获得55 916条蛋白序列, 以多肽序列格式“ fasta” 下载, 以实际抽取与汇报语言(perl)脚本检测数据库的完整性。数据库检索采用内置算法(Paragon)进行, 使用软件自带错误发现率(FDR)分析, P值由软件自动计算。具体参数(系统语言为英语, 下文采用英语加中文注释的方式)如下:Sample type(样本类型), Identification(鉴定); Cys alkylation(胱氨酸烷基化), Iodoacetic acid(碘乙酸); Digestion(酶解), Trypsin(胰酶); Instrument(仪器), TripleTOF 5600; Results quality(结果质控), Detected protein threshold(检测蛋白阈值)[Unsed ProtScore(conf)]> 0.05(10.0%)。取2个及以上肽段支持的蛋白, 去除1个肽段支持的蛋白鉴定到的蛋白经信号肽预测工具SignalP v4.0分析, 预测是否存在潜在的信号肽剪切位点及其位置, 并通过蛋白质亚细胞定位软件Prot Comp 9.0进行亚细胞定位预测。
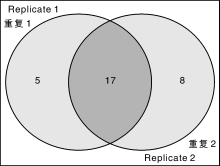
通过Protein Pilot 4.5软件分析, 2次重复分别采集到58 409张及45 759张图谱, 鉴定到的图谱分别是471张和387张, 对应的肽段分别是286条和241条。筛选2个肽段及以上的蛋白, 重复1鉴定到22个蛋白, 重复2鉴定到25个蛋白, 其中, 重复的有17个蛋白, 一共鉴定到30个蛋白(图1), 具体见表1。
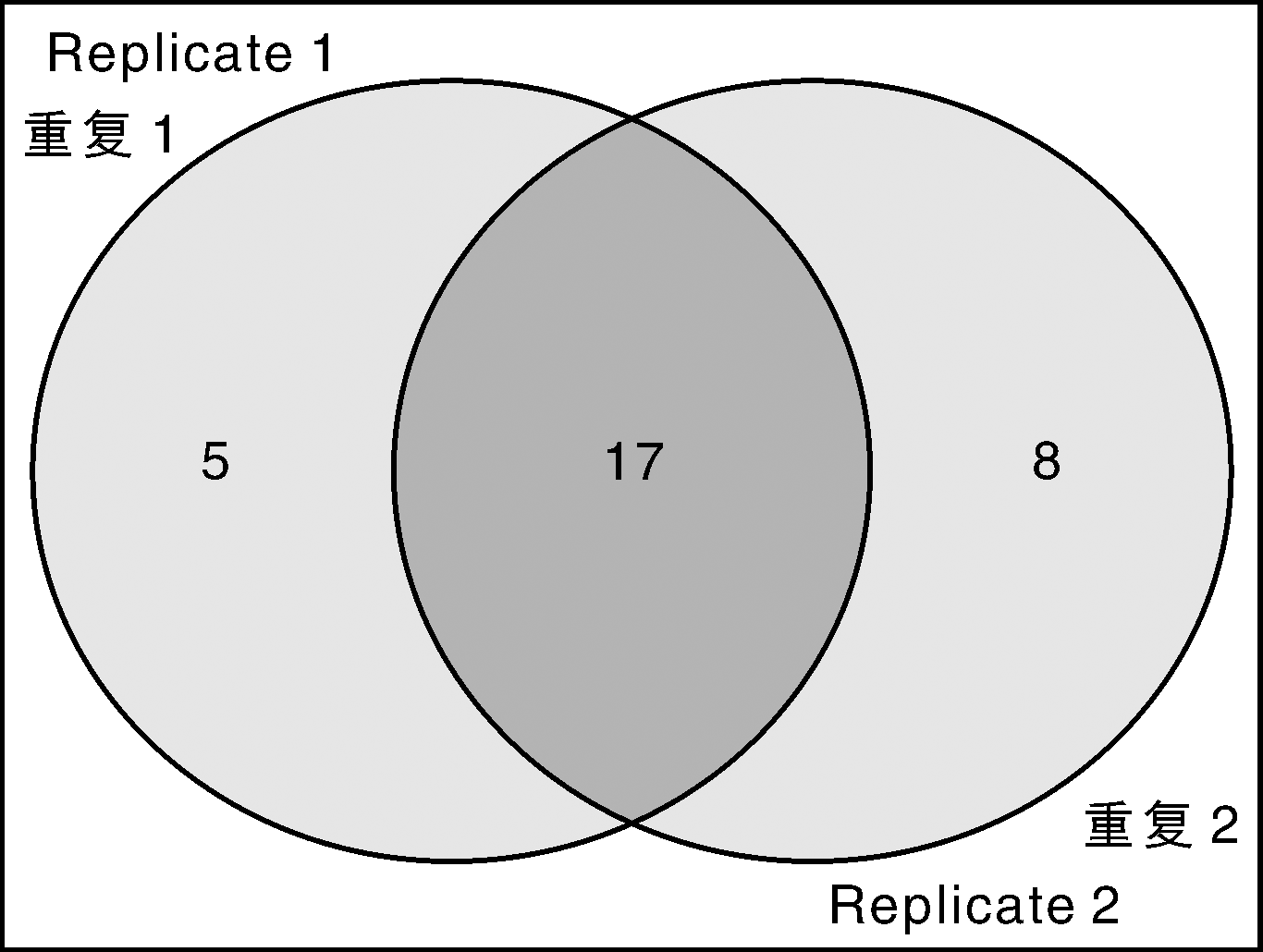 | 图1 鉴定结果Fig.1 Identified proteins from two independent replications |
| 表1 蛋白鉴定详细列表 Table 1 List of proteins identified |
鉴定到的蛋白经信号肽预测工具分析, 根据预测标准, 推测有24个蛋白含N端信号肽。进一步以软件分析蛋白的亚细胞定位(表2):26个蛋白分泌到胞外, 3个蛋白定位在细胞膜上, 只有1个蛋白定位在细胞质内。
| 表2 蛋白质生物信息分析结果 Table 2 Bioinformatics analysis of proteins |
对鉴定到的蛋白做进一步分析, 去除假设蛋白, 其中有11个蛋白是酶类, 根据功能进行分类, 主要集中在水解及能量代谢类别(表3)。其中, 部分酶在降解植物细胞壁成分中起重要作用:阿魏酸酯酶B, 可降解木质素, 木质素是植物细胞壁的重要组成部分; 糖苷转移酶(3-1, 3-葡聚糖酶), 可降解胼胝体, 胼胝体是由β -1, 3葡聚糖形成的多聚物, 分布在植物的胞间连丝和韧皮部。
| 表3 酶类分类表 Table 3 Classification of identified enzymes |
KEGG(Kyoto encyclopedia of genes and genomes, 京都基因与基因组百科全书)是系统分析基因功能、基因组信息的数据库。功能信息存在通路数据库中, 包括碳水化合物、氨基酸、核苷等的代谢及有机物的降解。KEGG不仅提供代谢途径, 还全面注解催化酶。对分泌蛋白进行KEGG代谢途径富集分析, 可以找到显著的通路, 有助于理解稻瘟病菌入侵水稻时的生物学调控通路。富集结果显示, 鉴定到的蛋白在果糖和甘露糖代谢、过氧物酶体、糖酵解途径、半乳糖代谢有富集(表4)。
| 表4 KEGG代谢途径富集分析结果 Table 4 KEGG enrichment of proteins identified |
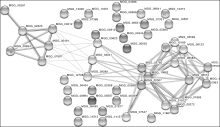
为了进一步挖掘分泌蛋白质的信息, 深入了解稻瘟病菌入侵水稻的分子机制, 对鉴定到的蛋白进行相互作用分析是有必要的。STRING数据库(https://string-db.org/)是一个基于已知和预测蛋白质相互作用的数据库, 这些相互作用包括蛋白质直接的(物理上的)和非直接(功能性的)相关性。其数据主要来源于基因组背景数据、高通量的实验数据、基因或者蛋白的共表达, 以及前人研究知识。分析结果如图2所示。在蛋白质网络分析中发现, MCG_09834与MCG_02625互作比较强, 进一步分析发现, MCG_09834是过氧化物酶2, 而MCG_02625是超氧化物歧化酶, 超氧化物歧化酶能将自由基催化成过氧化氢(H2O2), 而过氧化物酶是以H2O2为电子受体催化底物氧化的酶, 这两者间存在相互作用, 两者的结构和质谱鉴定见图3。
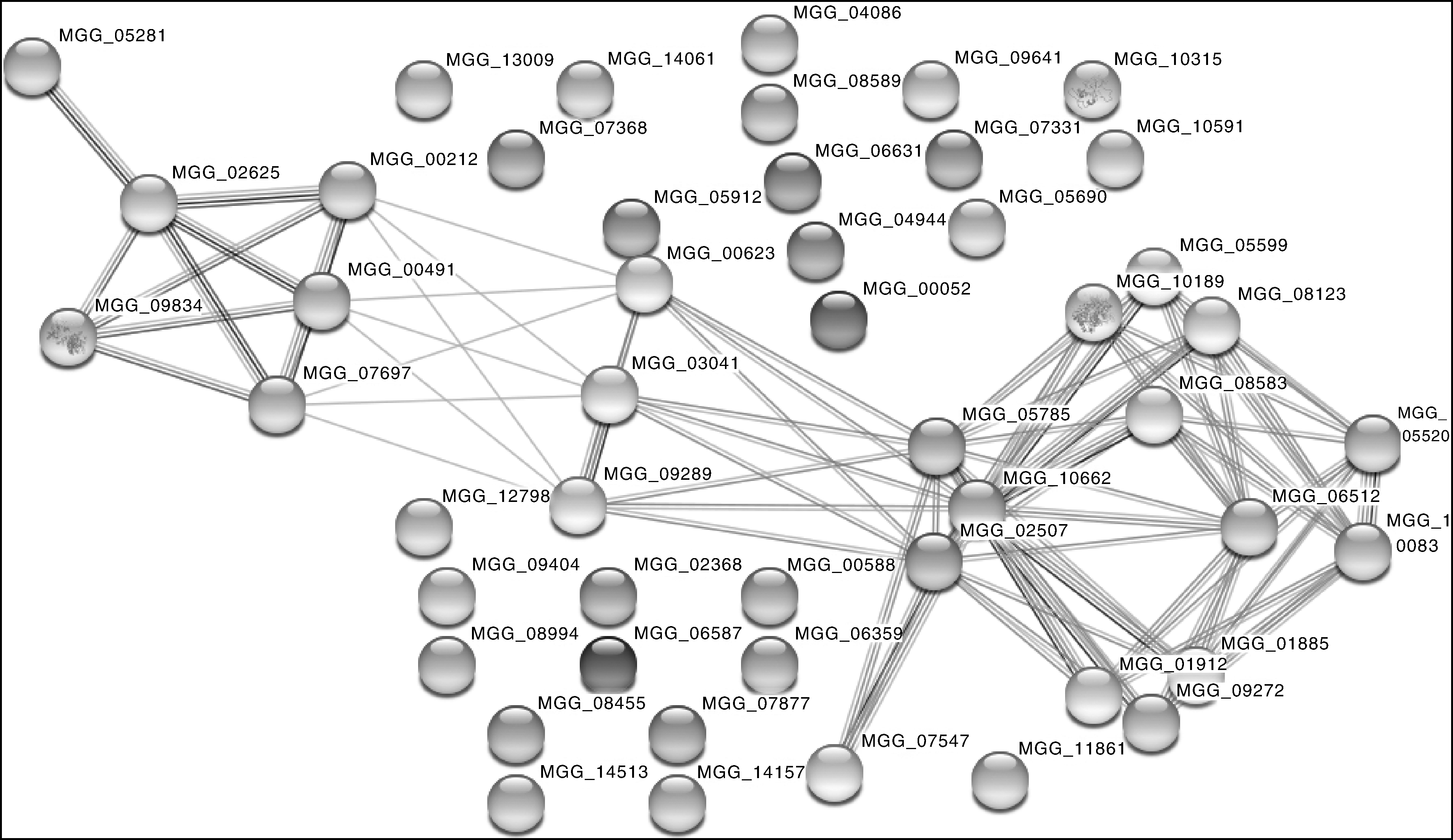 | 图2 STRING蛋白网络分析 圆圈代表蛋白质, 直线表示蛋白质之间的相互作用关系。Fig.2 STRING network analysis of proteins identified Balls indicated proteins, and lines indicated interactions between proteins. |
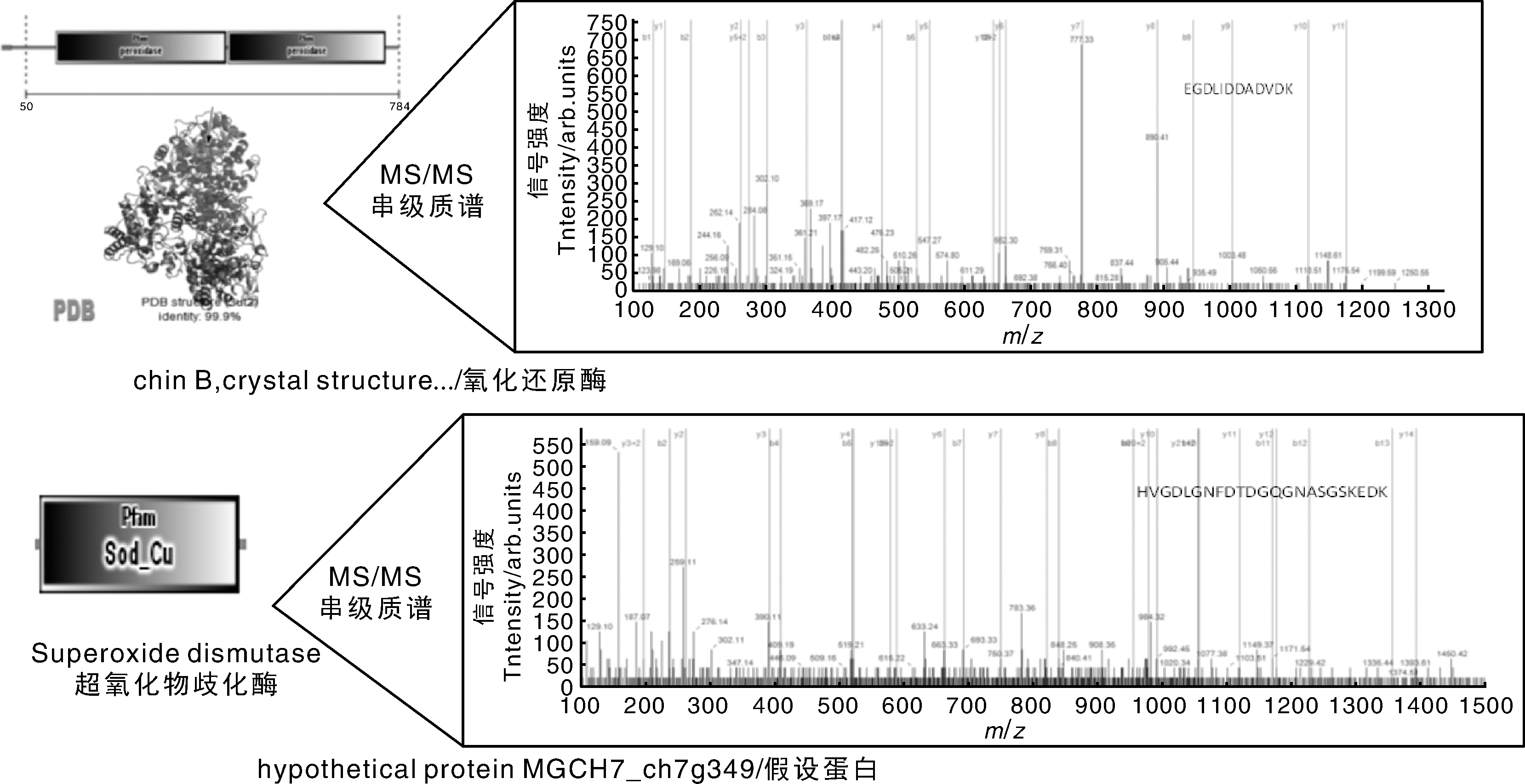 | 图3 酶结构(左)与质谱(右)Fig.3 Schematic diagrams of enzymes structures (left) and peptide fragments identified in mass spectrometer (right) |
分泌蛋白多为病原微生物与植物受体蛋白起作用的激发子和其他致病因子, 本研究通过LC-MS/MS鉴定稻瘟病菌的分泌蛋白质组, 并进行生物信息分析, 提示酶在稻瘟病菌入侵过程中的作用, 为进一步研究、揭示水稻与稻瘟病菌相互作用的分子机制奠定基础。
近年来, 随着质谱技术的快速发展, LC-MS/MS逐渐成为蛋白组学的主要研究技术。高效液相色谱(high performance liquid chromatography, HPLC)具备“ 三高” 特性, 即高自动化、高重现性和高分离效率, 成为肽段分离的重要方法, 结合质谱, 能够形成高自动化的分离和鉴定系统[17, 18]。在基于液质联用的蛋白质组学研究方法中, “ 鸟枪法” 最为常用, 即首先将蛋白质混合物酶解, 生成肽段混合物, 再将肽段混合物进行色谱分离, 在线用质谱检测洗脱的肽段[19]。本研究采用液质联用鉴定稻瘟病菌的分泌蛋白质组, 有30个蛋白被鉴定出, 与直接鉴定稻瘟病菌蛋白差别很大(数据未展示), 暗示稻瘟菌在开始入侵时, 可能仅分泌出少数蛋白来水解细胞壁, 入侵成功后, 再通过糖代谢提供能量, 做进一步侵染, 这可能是最有效的方式。
信号肽预测工具分析鉴定到的蛋白, 推测有24个蛋白含N 端信号肽。进一步分析亚细胞定位, 26个蛋白分泌到胞外, 3个蛋白定位在细胞膜上, 只有1个定位在细胞质内。这些分泌蛋白可分为两大类:一类是降解酶类, 包括水解酶、酯酶、肽酶、氧化还原酶等, 其中, 部分酯酶和氧化还原酶同时参与能量代谢; 另一类是结合蛋白, 包括环管kelch蛋白、CIA30家族蛋白, 这些蛋白帮助稻瘟病菌结合到细胞骨架和膜上。KEGG代谢途径富集分析也验证了酶类分类结果。这些蛋白在果糖和甘露糖代谢、过氧物酶体、糖酵解途径、半乳糖代谢途径有富集, 而这些代谢途径都与水解细胞壁、能量代谢相关。据此推测, 稻瘟病菌分泌环管kelch蛋白, 帮助稻瘟病菌结合到细胞壁上, 分泌1, 3-β -葡聚糖酶、β -葡萄糖苷酶, 并辅以肽酶及酯酶, 使细胞壁变得松弛, 阿魏酸酯酶B降解木质素, 从而降解水稻的纤维结构。β -葡聚糖酶和β -葡萄糖苷酶转移酶降解由β -1, 3 葡聚糖形成的多聚物(胼胝体), 再通过CIA30家族蛋白结合在膜上, 在肽酶、酯酶等水解酶帮助下, 稻瘟病菌进入细胞, 达到最终侵入目的。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|