


{kind=link}
{kind=link}
{kind=link}
{kind=link}
大麦4个穗部性状的关联分析
[胡倩文1  , 徐延浩
, 徐延浩1 , 王容1 , 张文英1 , 华为2, * , 吕超3 ]
, 徐延浩, 吕超
 |


作者简介:胡倩文(1993—),女,湖北荆州人,硕士研究生,主要从事植物分子育种研究。E-mail: 383008546@qq.com
穗部性状是大麦产量和品质的重要决定因素,良好的穗部结构应有一定的穗长基础、适合的小穗密度和每穗小穗数。以137份来源广泛的大麦材料为关联群体,利用224对SSR标记获取群体基因型,在2个环境试点下对穗粒数、小穗密度、小穗数、穗长进行性状观察,利用一般线性模型(GLM)和混合线性模型(MLM)两种模型进行4个穗部性状与SSR标记的关联分析。供试大麦群体的4个穗部性状差异显著,穗粒数与小穗数、穗长与小穗密度显著相关。224对SSR标记共检测出479个等位变异,平均有效等位基因数、Shannon's指数和PIC分别为1.765、0.612和0.327。供试材料的遗传相似系数(GS)变化范围为0.486~0.891,并在GS值为0.580时,将材料分为2个类群。群体结构分析也将供试材料分为2个亚群。GLM分析显示,与穗粒数、小穗密度、小穗数、穗长相关联的标记位点数量分别有33、13、34、6,这些标记遍布大麦7条染色体,单个标记对表型变异的解释率为8.65%~50.08%( P<0.001);MLM分析显示,与上述4个性状相关联的标记位点数量分别为4、3、4、1,这些标记位于大麦1H、2H和4H染色体,单个标记对表型变异的解释率为3.12%~16.95%( P<0.001)。 Ind1003、 Ind2030、 Ind2055、 Ind4012这4个标记在2个环境中都能被2种模型检测到,且同时与穗粒数和小穗数相关联。
Spike traits are important determinants of barley yield and quality. A good spike structure should have a certain spike length, suitable spikelet density and spikelet number per spike. In this study, 137 barley materials from a wide range were used as the population for association analysis. A total of 224 pairs of SSR markers were used to analyze the genotype of the population. Under two environmental experiments, grain number per spike, spikelet density, spikelet number per spike and spike length were recorded. General linear model (GLM) and mixed linear model (MLM) models were used to analyze the association between four spike traits and SSR markers. The results showed that there were significant differences among the four spike traits of the tested barley population. Grain number per spike was significantly related to spikelet number per spike, and spike length was significantly related to spikelet density. A total of 479 alleles were detected for SSR markers. The average effective allele number, Shannon's index and PIC were 1.765, 0.612 and 0.327, respectively. The genetic similarity coefficient (GS) of the tested materials ranged from 0.486 to 0.891. The barley materials were divided into two groups at GS value level of 0.580. Population structure analysis also divided the materials into two subgroups. The results of GLM analysis showed that there were 33, 13, 34 and 6 loci associated with grain number per spike, spikelet density, spikelet number per spike and spike length, respectively, and the phenotypic variation explained by a single marker ranged from 8.65% to 50.08% ( P<0.001). These markers were located on seven chromosomes of barley. The results of MLM analysis showed that there were 4, 3, 4 and 1 marker sites associated with the four spike traits described above, respectively. The phenotypic variation explained by a single marker ranged from 3.12% to 16.95% ( P<0.001). These markers were located on the chromosome 1H, 2H and 4H of barley. The four markers Ind1003, Ind2030, Ind2055 and Ind4012, which associated to both grain number per spike and spikelet number per spike, could be detected by two models in two tests.
大麦是世界第四大谷物, 主要用于饲料和酿造[1]。穗粒数、小穗密度、小穗数、穗长是影响大麦产量和品质的重要穗部性状。穗粒数是产量三要素之一, 合适的小穗密度是决定大麦籽粒饱满度和麦芽品质的重要因素[2, 3], 因此, 研究控制大麦穗部性状的基因位点是大麦育种工作的重要方向。
大麦长、短穗与稀、密穗均受一对基因控制, 且长穗和稀穗表现为显性[4]。研究者发现, 大麦籽粒密度的变化是由microRNA172与其在转录因子HvAP2的mRNA相应结合位点互相作用的结果[5]。大麦的密穗性状被定位于7H染色体0.37 cM的区间内[6]。已有研究报道了穗长等4个穗部性状的相关QTLs, Wang等[2]采用连锁分析在2H和3H染色体上检测到2个控制穗长和籽粒密度的QTL, 并在2H染色体18.9 cM和25.6 cM处检测到2个穗粒数QTL; Baghizadeh等[7]共检测到位于4号连锁群上的3个穗长和位于1号连锁群的3个小穗数的QTL位点; Wang等[8]检测到3个穗长、4个小穗密度、2个穗粒数和6个小穗数的QTL位点。少有研究进一步鉴定大麦小穗数QTLs及其与穗粒数等穗部性状的遗传关联性。
关联分析(association analysis)是一种以连锁不平衡为基础, 检测自然群体内目标性状与遗传标记或者候选基因相关关系的分析方法。关联分析具有材料遗传基础广泛等优点[9], 已在水稻[10]、小麦[11]、大麦[12]等作物中挖掘了多个优异等位基因。司二静等[13]、赖勇等[14]、孟亚雄等[15]运用关联分析方法, 检测到多个与大麦穗长、穗粒数、小穗密度相关的遗传位点。但这些关于大麦穗部性状的关联分析在材料数量、标记数量、环境试点都较为有限[13, 14, 15]。
多种环境试验通常用于评估基因型的性能, 一些QTL对环境敏感, 在不同的环境可能有不同的影响, 在多个环境下鉴定产量性状的稳定QTL, 对于将其应用于标记辅助选择至关重要。本研究以137份来源比较广泛的大麦材料为关联群体, 利用224对SSR标记对群体进行基因型分析, 并在2个环境试点下对穗粒数、小穗密度、小穗数、穗长进行关联分析, 以期为大麦穗部性状的遗传研究和分子标记辅助育种提供更多的信息。
供试137份大麦材料包含56份中国栽培材料、52份国外栽培材料、29份西藏半野生大麦(表1)。2017年11月上旬分别将137份材料种植于长江大学太湖(TH)试验基地和荆州农业科学院(JN)试验基地。采用完全随机区组设计, 每试验点设置2个重复, 单个材料每个重复种植3行, 行长2.0 m、行距0.25 m、株距8.0 cm, 田间管理同常规。每个重复取中间行长势一致的5个单株考察穗粒数(grain number per spike, GNS)、穗密度(spikelet density, SD)、小穗数(spikelet number per spike, SNS)、穗长(spike length, SL)。小穗密度=小穗排数/穗长。利用SPSS 19.0软件进行各个环境下穗部性状的统计和Pearson相关性分析。
| 表1 供试大麦材料的名称和来源 Table 1 Name and origin of barley accessions used in this study |
取三叶期新鲜幼嫩叶片, 用CTAB法提取基因组DNA。用NanoDrop2000c超微量分光光度计(Thermo, 美国)测定DNA浓度。本试验中224对SSR标记引物序列及其染色体位置信息由澳大利亚默多克大学李承道教授提供。引物由南京金斯瑞生物科技有限公司合成。
PCR反应体系的试剂均购自上海博彩生物科技有限公司, 扩增体系(10 μ L)包含1× buffer, Mg2+ 0.2 mmol· L-1, dNTPs 0.2 mmol· L-1, Taq酶0.75 U· L-1, 上下游引物各5 μ mol· L-1, 模板DNA 50 ng。PCR扩增程序:95 ℃预变性4 min; 94 ℃变性30 s, 54~61 ℃退火30 s, 72 ℃延伸45 s, 33个循环; 72 ℃延伸10 min。PCR产物用6%的非变性聚丙烯酰胺凝胶电泳分离, 银染显色[16]。以二进位制来统计PCR扩增产物的电泳结果, 在同一电泳迁移位置上, 有DNA扩增条带记为“ 1” , 无条带记为“ 0” 。
通过Popgene 32软件统计分析观测等位基因数(observed number of alleles, Na)、有效等位基因数(effective number of alleles, Ne)、观察杂合度(observed heterozygosity, Ho)、期望杂合度(expected heterozygosity, He)、Shannon's信息指数(Shannon's Information index, I)及Nei's基因多样性指数(Nei's gene diversity index, H)。利用PIC_CALC 0.6计算SSR位点多态性信息含量(polymorphism information content, PIC)。以NTSYS-pc计算遗传相似性系数(genetic similarity coefficient, GS), 按照非加权平均法(unweighted pair-group method with arithmetic means, UPGMA)和SHAN程序对自然群体材料进行聚类分析。
以Structure 2. 3. 4软件进行贝叶斯聚类, 分析群体的遗传结构并确定最佳的群体分组(K)。K值的取值范围为1~10, 将MCMC(Markov Chain Monte Carlo)开始时的不作数迭代(length of burn-in period)设为10 000次, 再将不作数迭代后的MCMC设为100 000次, 迭代次数(number of iterations)设置为8, 参照Evanno等[17]的方法计算Δ K, 确定合适的K值, 并计算Q参数。
运用Tassel 2.1软件一般线性模型(general linear model, GLM)和混合线性模型(mixed linear model, MLM)进行关联分析。GLM分析将Q作为协变量进行回归分析; MLM分析采用Q+K方法, 分析方法选择EM。利用 4个穗部性状表型数据对分子标记逐一进行回归分析, 当P< 0.001时认为该标记与目标性状关联, 并计算标记对表型变异的解释率。利用MapChart 2.32通过遗传距离绘制关联位点在染色体上的分布。根据关联分析结果, 通过R软件CMplot包(https://github.com/YinLiLin/R-CMplot)绘制曼哈顿图。
在分析的大麦穗部性状中(表2), 穗粒数和小穗数的变异幅度比较大, 穗粒数在JN和TH两试点的分布分别为15.000~89.333和20.000~93.333, 变异系数分别为50.997%和50.484%, 小穗数在JN和TH两环境试点的分布分别为20.000~92.000和21.333~98.000, 变异系数分别为49.211%和50.188%, 这可能是因为把二棱和六棱大麦混合统计的原因[7]。小穗密度和穗长变异幅度比较小, 小穗密度在JN和TH的分布分别为2.220~6.173和2.273~5.180, 变异系数分别为21.509%和20.045%。穗长在JN和TH两环境试点的分布分别为3.867~12.300和5.167~13.500, 变异系数分别为21.723%和18.392%。
| 表2 两试点大麦材料4个穗部性状的变异 Table 2 Variations of 4 spike traits of barley at two test sites |
穗粒数与小穗数在2个环境中均呈极显著正相关, Pearson相关性系数在JN和TH两个试点分别为0.970和0.997, 具有较高的遗传关联性。穗长与小穗密度在2个环境试点均达极显著负相关。穗粒数与穗长、小穗密度, 小穗数与小穗密度、穗长在2试点均无显著相关性(表3)。
| 表3 四个大麦穗部性状在两试点的相关性分析 Table 3 Correlation analysis of 4 spike traits of barley at two test sites |
224对SSR标记在137份大麦材料中共检测到479条多态性条带, 平均等位变异为2.138, 平均有效等位基因数为1.765± 0.364, 范围为1.058~2.990; Shannon's信息指数平均值为0.612± 0.184, 范围为0.129~1.104; 多态性信息含量平均值为0.327± 0.101, 范围为0.054~0.592; 期望杂合度平均值为0.409± 0.131, 范围为0.055~0.668; Nei's基因多样性指数平均值为0.408± 0.130, 范围为0.055~0.666(表4)。
| 表4 SSR标记在137份大麦材料中的多态性分析 Table 4 Polymorphism analysis of SSR markers in 137 barley materials |
供试137份大麦材料的平均遗传相似系数(GS)为0.613, 变幅为0.486~0.891。其中鄂大麦8号与创43的GS值均达到最大值0.891; 1592与1588的GS值达到0.887。Ibon-w08-9与80284的GS值最小0.486; 其次08PJ-37青与1580 GS值达到0.493。
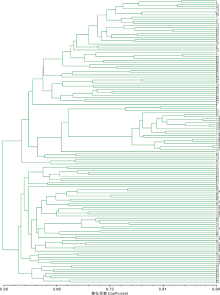
利用NTSYS-pc进行聚类分析, 根据UPGMA法构建了遗传关系聚类图(图1), 结果表明, 在GS值为0.580水平时, 137份大麦材料被分为了2大类群, 第1类群包含大部分澳大利亚品种和国内湖北品种等共80份材料; 第2类群包含供试材料中所有的云南品种以及国内北方品种等57份大麦种质材料。聚类结果中, 西藏半野生大麦材料没有单独聚为一类, 第1类群包含17份西藏野生大麦材料, 第2类群包含12份西藏野生大麦材料。
通过对137份大麦材料的群体结构分析, 贝叶斯方法结果显示, 随着K值的增大, lnP(D)值整体呈增大趋势, 无明显拐点, 无法直接确定合理的K值(图2-A), 因此, 参照王国荣等[16]的方法计算Δ K确定合适的K值(图2-B), 在K=2时得到Δ K的最大值314.332, 将137份大麦材料分成2个亚群, 并绘制供试材料群体结构(图2-C), 2个亚群分别包括80、57份材料, 此结果与聚类分析结果一致。137份材料的Q值均大于0.7。表明这些大麦材料遗传背景简单, 亚类间缺少基因交流。
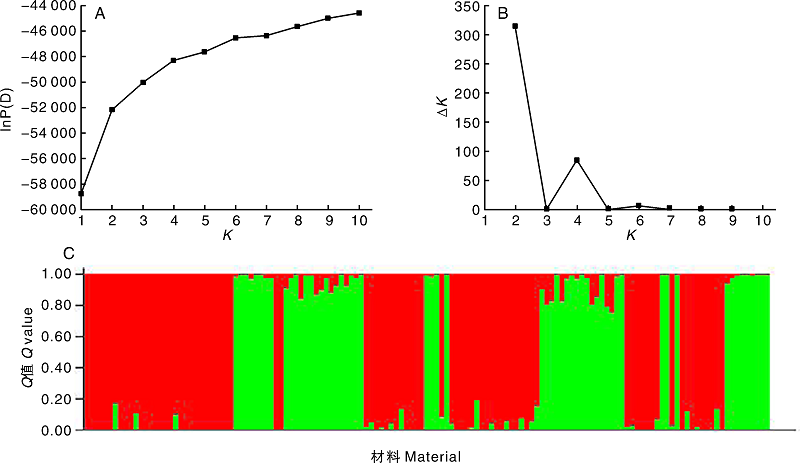 | 图2 基于SSR标记的137份大麦材料群体遗传结构分析 A, lnP(D)值随K值变化折线图; B, Δ K值随K值变化折线图; C, 群体遗传结构图(红色代表亚群1, 绿色代表亚群2)。Fig.2 Population genetic structure of 137 barley material based on SSR markers A, Line chart of lnP(D) with change of K-value; B, Line chart of Δ K with change of K-value; C, Chart of population genetic structure (the red part represented subgroup 1, and the green part represented subgroup 2). |
GLM分析共检测到33个与穗粒数相关联的标记位点, 其中有20个位点在2个环境中均被检测到(表5、图4), 这20个标记的表型变异解释率为11.14%(Ind3013)~48.89%(Ind4012)。MLM分析结果共检测到4个(Ind1003、Ind2030、Ind2055、Ind4012)与穗粒数相关联的位点, 且这4个位点在2个环境试点均被检测到, 其中位点Ind4012的表型变异解释率最高(16.95%, JN试点)。
| 表5 关联标记及其对表型变异的解释率 Table 5 Associated markers and the explained phenotypic variation % |
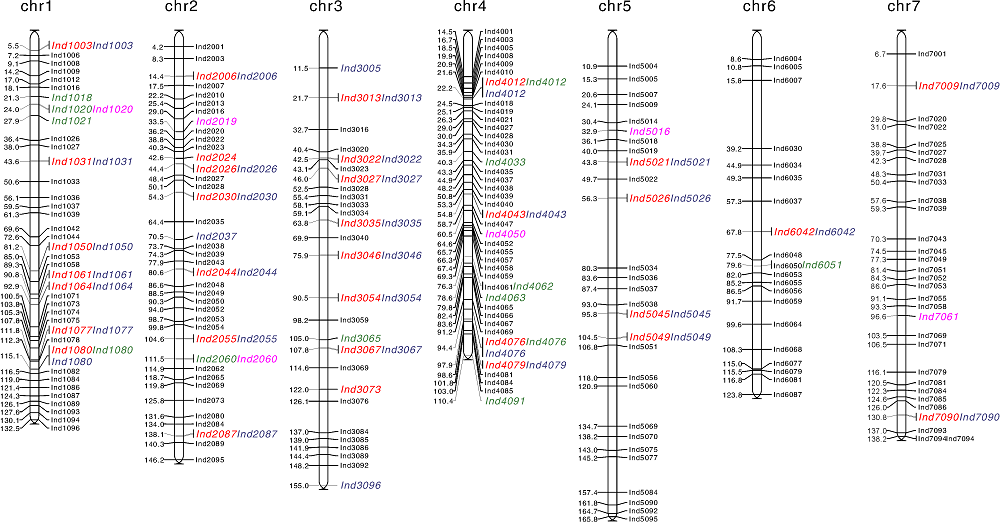 | 图3 SSR标记在染色体上的分布图及与性状关联的位点分布图 红色, 与穗粒数关联位点; 绿色, 与小穗密度关联位点; 蓝色, 与小穗数关联位点; 紫色, 与穗长关联位点。Fig.3 Distribution of SSR markers on chromosomes and loci associated with traits Red, Loci associated with grain number per spike; Green, Loci associated with spikelet density; Blue, Loci associated with spikelet number per spike; Purple, Loci associated with spike length. |
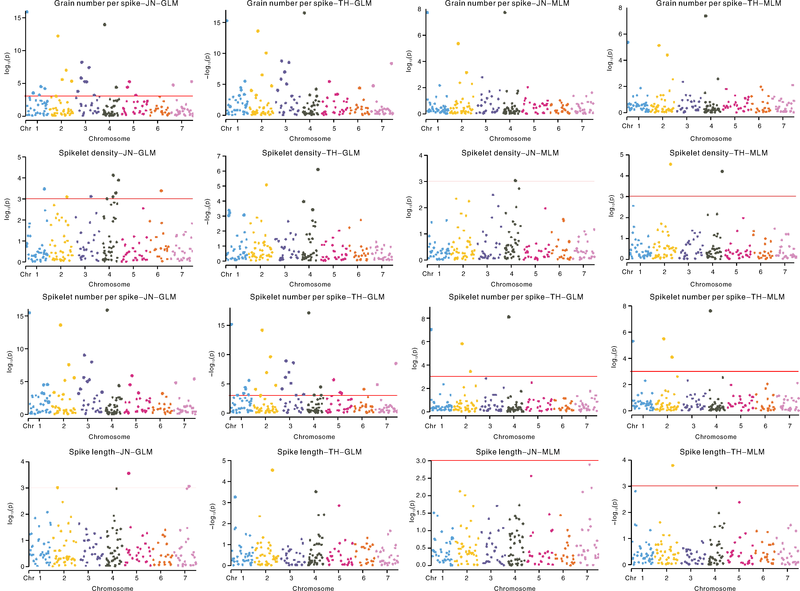 | 图4 两种模型下两个试点的穗粒数、小穗密度、小穗数、穗长关联分析曼哈顿图Fig.4 Manhattan of association analysis by two models for grain number per spike, spikelet density, spikelet number per spike, spike length from two test points |
GLM分析结果共检测到13个与小穗密度相关联位点, 其中有4个位点在2个环境中均被检测到(表5和图4), 这4个位点在2个环境的表型变异解释率为9.03%(Ind2060)~21.48%(Ind4091)。MLM分析结果共检测到3个与小穗密度相关联的位点。
GLM分析结果共检测到34个与小穗数相关联位点, 其中有24个在2个环境试点均能被检测到(表5和图4), 这24个标记位点的表型变异解释率为11.37%(Ind2055)~50.08%(Ind4012)。
MLM分析结果共检测到4个与小穗数相关联的位点, 且这4个位点在2个环境试点均被检测到, 其中位点Ind4012的表型变异解释率最高(14.46%, JN试点)。
GLM分析结果共检测到6个标记与穗长相关联的标记位点, MLM分析结果仅检测到1个标记与穗长相关联。
MLM分析中检测到的与穗部性状关联的位点均在GLM分析中被检测到。本研究共检测到与穗粒数、小穗密度、小穗数、穗长相关联的位点分别为33、13、34、6个。同时与穗粒数和小穗数、小穗密度和穗长相关联的位点分别为28、2个。标记位点Ind1080、Ind4012和Ind4076同时与穗粒数、小穗密度、小穗数相关联(表5、图3)。
穗粒数、小穗密度、小穗数、穗长为多基因控制的复杂性状[3, 8, 18]。本研究分析的大麦穗部性状中, 穗粒数的变异幅度最大, 在2试点的变幅分别为15.000~89.333和20.000~93.333, 这可能是因为把二棱和六棱大麦混合统计的原因。李静烨等[3]研究发现, 大麦小穗数与穗粒数高度相关, 并检测出紧密遗传连锁的QTLs。本研究也发现小穗数与穗粒数呈极显著正相关, 并检测到28个同时控制穗粒数和小穗数的关联位点, 进一步证实了小穗数与穗粒数的遗传关联性。
种质资源材料是进行育种的基础, 利用分子标记技术能有效地分析不同材料间的遗传差异, 为后期亲本杂交的组合配置提供一定的参考。此前, 王国荣等[16]、赖勇等[19]和孟亚雄等[20]都发现国内青稞以及大麦育种材料间的亲缘关系较近。赖勇等[19]研究表明, 88份青藏高原青稞材料的平均GS值为0.761, 变幅为0.497~0.970; 孟亚雄等[20]研究表明, 89份来源不同大麦材料的GS值为0.520~0.873。本研究137份大麦群体来源于国内13个省和11个其他国家, 平均GS值为0.613, 变幅为0.486~0.891。与前人的研究相比[19, 20], 本研究大麦群体GS值相对更小, 材料之间的亲缘关系相对更远, 遗传基础相对更丰富, 说明后续大麦育种材料的选择应加强注意外来种质材料的引进。
本研究采用一年两点试验, 利用GLM和MLM两种模型对标记与表型进行关联分析, 寻找不同环境中被检测出的标记位点, 发现大麦7条染色体上都分布有与穗粒数相关联的位点, 这与前人研究结果一致[2, 8, 13, 15, 21, 22]。在本研究检测的33个穗粒数相关位点中, 有9个位点Ind1031、Ind1077、Ind2006、Ind2044、Ind2087、Ind3013、Ind3035、Ind5026和Ind7009分别与司二静等[13]、孟亚雄等[15]、赖勇等[23]、Wang等[8]、Pillen等[22]已报道的位点相近, 其中Ind2044(2H:80.6 cM)和Ind2087(2H:138.1 cM)分别与Wang等[8]qGs2-2(2H:85.91 cM)和qGs2-4(2H:128.71 cM)位置相近; Ind5026(5H:56.3 cM)与Pillen等[22]检测的QTL位点Bmag0113(5H:61 cM)位置相近。除相近的9个位点外, 其余24个位点前人均未报道过, 其中4个新的穗粒数相关位点在两个试点均被检测到, 值得关注, 其中Ind1003位于基因HORVU1Hr1G066030区间内, 该基因的编码蛋白为含有PlsC结构域的蛋白质, 参与磷脂酰肌醇酰基链重塑, 影响转移酶活性, 参与内膜系统整体组成; Ind2030位于基因HORVU2Hr1G046850区间内, 该基因的编码蛋白为含RIO结构域的蛋白质, 参与蛋白质磷酸化, 影响ATP结合, 影响蛋白丝氨酸或苏氨酸激酶活性。
关于小穗密度的研究较少, 前人在2H、3H、4H、6H、7H染色体上都检测到控制小穗密度的位点[2, 6, 8, 23], 本研究在1H染色体上检测到了多个关联位点, 但在7H染色体上没有检测到位点。在本研究检测到的13个与小穗密度相关联位点中, 仅一个位点Ind6051与前人研究位点相近, 位点Ind6051(6H:79.6 cM)与赖勇等[23]检测得到的HVM31(6H:72.8 cM)位置相近。其余12个位点前人均未报道过, 其中2种分析模型均能检测到Ind4063(chr4H:595106302-595106382), 值得关注, 其位点上有一个基因HORVU4Hr1G074700, 该基因参与花粉外壁形成和木栓质的生物合成过程, 影响脂肪酰基辅酶A还原酶的活性, 参与叶绿体的形成。
已报道的小穗数的位点位于1H、2H、4H[8, 24], 本研究在7条染色体上均检测到位点, 一共检测到34个位点与小穗数相关联。有2个位点Ind2037和Ind2044与前人研究位点相近, 位点Ind2037(2H:70.5 cM)和Ind2044(2H:80.6 cM)与Wang等[8]检测到qSms2-1(2H:69.01 cM)和qSms2-4(2H:79.01 cM)位置相近, 同时Ind2044(2H:652.41 Mb)与Hu等[24]检测到的表型变异解释率为65.07%~90.41%的qtnSMS-2H-9(2H:652.03 Mb)重叠。
与前人研究相比[8, 14, 21, 24, 25, 26], 本研究在3H和6H没有检测到穗长相关位点。本研究共检测到6个与穗长相关的位点, 有2个位点Ind2060和Ind7061与前人研究位点相近, 其中Ind2060(2H:111.5 cM)位于Hori等[25]检测到的穗长QTL(vrs1-Bmag125, 2H:82.4~122 cM)区间内, 且该位点与司二静等[13]检测到的Bmag125(2H:122 cM)位置接近。其余4个位点前人均未报道过, 其中位点Ind4050(chr4H:550655515-550655625)附近有一个基因HORVU4Hr1G066090, 该基因参与脂质代谢过程, 影响O-酰基转移酶活性, 参与膜的整体组成。
本研究利用GLM和MLM两种模型对JN和TH两个环境试点下大麦的穗粒数、小穗密度、小穗数和穗长进行关联分析, 获得多个能在2个环境同时检测到的标记位点, 其中多个标记位点与前人研究结果一致, 表明本研究方案和材料可以用于相关基因的检测研究; 同时, 挖掘到多个未被报道地潜在基因位点, 为后续大麦穗部性状的遗传研究和分子标记辅助选择育种提供重要参考。
(责任编辑 张 韵)
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|